
Лесли хорнби и синдром марфана
Синдром Марфана – редкая генетическая болезнь, которая плохо поддается лечению, характеризуется аномалиями соединительной ткани и проявляется в нарушениях зрения, опорно-двигательного аппарата, сердечно-сосудистой системы и других симптомах.
Среди внешних проявлений у больных синдромом Марфана, который также называют «синдромом гениев», отмечаются непропорционально длинные руки, кисти и стопы, а также пальцы рук и ног. В материале 24СМИ – российские и зарубежные знаменитые люди с синдромом Марфана.
1. Никколо Паганини
Знаменитый композитор и скрипач стал жертвой генетической болезни. Длинные необыкновенно подвижные и гибкие пальцы позволили музыканту добиться успеха в выбранной профессии.
Внешность Никколо Паганини, по описаниям современников, выглядела странной и пугающей: чрезмерная худоба и бледность, высокий рост, неправильная осанка, деформированная грудная клетка, ноги неодинаковой длины и непропорциональность. К тому же гениальный музыкант на своих концертах и выступлениях поражал публику необычной манерой исполнения, лихорадочностью движений, которые сделали Паганини легендой.
2. Ганс Христиан Андерсен
Известный во всем мире детский писатель Ганс Христиан Андерсен, судя по описаниям современников, тоже страдал синдромом Марфана. Внешность Андерсена выглядела на редкость нескладной, странной и даже пугающей: высокий рост, худощавость, угловатость, непропорционально длинные руки и ноги. Размер обуви писателя также был огромен, даже для его роста. При этом у Андерсена был заметно выступающий вперед нос римской формы, высокий лоб и необычайно тонкие губы.
3. Авраам Линкольн
Знаменитые люди с синдромом Марфана встречаются и в мире политики. Самый известный государственный деятель, у которого диагностировали эту болезнь, – Авраам Линкольн, 16-й президент США. Рост политика составлял 193 см, а телосложение было типичным для генетического заболевания – у Линкольна наблюдалась худощавость и нескладность, длинные ступни и кисти рук, гибкие пальцы и узкая грудная клетка.
4. Корней Чуковский
Исследователи считают, что известный русский детский писатель Корней Иванович Чуковский также страдал генетической болезнью. Современники описывали его как человека с длинными руками и ногами, а также большим носом.
Портрет Чуковского мог легко стать прототипом милиционера дяди Степы из стихотворения Сергея Михалкова. К тому же Корней Иванович отличался высокой работоспособностью, которую объясняют повышенным содержанием гормона адреналина в крови, вызванным синдромом Марфана.
5. Майкл Фелпс
В мире спорта также существуют знаменитые люди с синдромом Марфана. Американский пловец Майкл Фред Фелпс родился в 1985 году и за свою спортивную карьеру стал 23-кратным олимпийским чемпионом, рекордсменом и обладателем 28 олимпийских наград.
Сам пловец отрицает указанный диагноз, однако о наличии болезни говорят симптомы, заметные невооруженным глазом. Голеностопный сустав спортсмена наделен удивительной эластичностью: Майкл умеет выгибать ступню сильнее, чем балерины. Известно также, что спортсмен носит 47-й размер обуви, что превышает среднестатистические показатели для людей его роста, а размах его рук составляет 203 см, при этом рост Майкла – 193 см.
6. Юлий Цезарь
Некоторые исследователи сходятся во мнении, что в подборку «Знаменитые люди с синдромом Марфана» следует включить и великого древнеримского полководца Юлия Цезаря. На наличие заболевания указывают изображения и описания его личности, где отмечается выраженная худоба, бледность кожи, высокий рост и длинные пальцы рук. Кроме того, предполагается, что Цезарь страдал головными болями, эпилепсией и болезнями сердца.
7. Шарль де Голль
Знаменитый французский военный и государственный деятель Шарль де Голль также страдал редкой болезнью. Политик отличался высоким ростом, который, однако, не был заметен в сидячем положении, что говорит о непропорциональности тела. Также де Голль отличался узкими плечами, длинными конечностями и узким вытянутым лицом.
Эти внешние признаки политик унаследовал от отца, и некоторые исследователи осмелились сделать предположение, что знаменитый француз страдал синдромом Марфана. К тому же Шарль де Голль при такой нескладной конституции вел активную военную жизнь и обладал незаурядным интеллектом и удивительной проницательностью.
Источник

Если из-за генетической аномалии во время внутриутробного развития нарушается формирование соединительной ткани, то дети рождаются с синдромом Марфана. В структуре коллагеновых волокон возникает дефект, что проявляется поражением костей, связочного аппарата, сердца, глаз. Наиболее опасным осложнением является расслаивающая аневризма аорты. Она может быть причиной смертельного исхода, поэтому пациенты должны постоянно наблюдаться кардиологом и кардиохирургом.
Известные люди с синдромом Марфана
Синдром Марфана назван по имени педиатра, который наблюдал девочку с этим заболеванием на протяжении 20 лет. Имеется много интересных фактов о людях, имеющих характерные признаки патологии. Первая манекенщица (Лесли Хорнби – «Твигги»), которая была прототипом для всех чрезмерно худых моделей, болела синдромом Марфана. Наиболее известные личности, о которых есть подобные сведения:
- президент А. Линкольн,
- скрипач Н. Паганини,
- писатель Г. Х. Андерсен,
- композитор С. Рахманинов,
- олимпийский чемпион М. Фелпс.
Распространенность симптомокомплекса у многих одаренных людей дала возможность предположить, что он придает экстраординарные способности.
Рекомендуем прочитать статью об аортальном пороке сердца. Из нее вы узнаете о распространенности заболевания и причинах его развития, симптомах патологии, диагностике и лечении.
А здесь подробнее о симптомах аневризмы аорты.
Причины заболевания
Болезнь связана с генетической аномалией, которая передается по наследству или возникает спонтанно. Мутациям подвергается часть хромосомы, которая отвечает за образование фибриллина. Этот белок делает соединительную ткань прочной и упругой. При его дефиците страдают структуры, которые имеют наибольшее количество подобных волокон:
- сосуды, особенно аорта;
- сухожилия и связки, в том числе хрусталика глаза;
- подкожная клетчатка.
Так как изменения генов могут быть разнообразными (изучено более 100 вариантов), а также они комбинируются с другими патологиями строения хромосомного аппарата, то проявлена синдрома Марфана отличаются.
Классификация заболевания
Встречаются легкие случаи и крайне тяжелые, с быстрой прогрессией нарушения деятельности сердечно-сосудистой, легочной системы, почек. Промежуточное положение между ними занимает синдром Марфана средней тяжести.
Также существует подразделение этого заболевания на клинические формы по наличию поражения систем организма:
- стертая – патология в одной или двух системах со слабыми отклонениями от нормы;
- выраженная – незначительные нарушения в 3 системах или серьезные в 1;
- тяжелая – прогрессирующее нарушение деятельности 3 систем.
Стабильным вариантом течения считается болезнь при наличии изменений в работе или строении органов, которые не прогрессируют на протяжении более 1 года.
Признаки наличия отклонения у детей и взрослых
Проявления патологии многообразны, и их первые признаки могут быть обнаружены при рождении ребенка или постепенно нарастают во взрослом возрасте.
Наиболее тяжело протекает классическая форма, при которой изменения имеются в самом раннем периоде развития.
К ним относятся такие симптомы:
- длина тела при рождении более 53 см, у взрослых достигает 190 см (у женщин 175 — 180);
- туловище короткое, а конечности длинные с тонкими пальцами, паукообразной формы;
- мышцы и подкожная клетчатка развиты слабо;
- небо высокое по типу арки, нос длинный, прикус нарушен;
- суставы с избыточной подвижностью;
- имеются вывихи шейного отдела, искривление позвоночника;
- грудная клетка воронкообразной формы;
- плоскостопие;
- головка бедренной кости вдавливается вглубь таза.
Наиболее опасными являются не внешние дефекты, придающие пациентам характерный вид, а патология строения сердца и сосудов. Именно эти пороки развития определяют продолжительность жизни больных и занимают ведущее место в клинической картине. Наиболее типичные из них:
- стеноз магистральных сосудов – аорты и легочной артерии;
- отверстия в перегородках сердца;
- аортальная недостаточность, расширение восходящего сегмента и аневризмы аорты;
- удлиненные створки митрального клапана с разрывом сухожилий, которые их прикрепляют.
Стеноз аорты при синдроме Марфана
Из-за поражений сосудов и строения клапанного аппарата пациенты страдают от недостаточности кровообращения, они подвержены развитию бактериального эндокардита, аритмии в виде тахикардии, мерцания или трепетания предсердий и желудочков. При комбинированных функциональных и анатомических нарушениях смертельный исход может быть на первом году жизни ребенка.
Нарушения зрения выявляются в виде двустороннего вывиха хрусталика, они, как правило, нарастают до 5 лет и сопровождаются потерей зрения. Также при синдроме Марфана возникают такие офтальмологические патологии:
- близорукость,
- увеличенная и более плоская роговица по сравнению с нормой,
- недоразвитая радужная оболочка,
- косоглазие,
- деформированные сосуды сетчатки.
При нарушении развития твердой оболочки мозга в поясничном отделе позвоночника возникает выпячивание содержимого спинного-мозгового канала, что сопровождается болью и онемением нижних конечностей. В легких возможно формирование эмфиземы, пневмоторакса, почки и мочевой пузырь, а у женщин и матка смещаются книзу, часто происходит растяжение связок, образование паховых и бедренных грыж.
По уровню умственного развития пациенты не отличаются от здоровых людей, а в некоторых случаях имеют очень высокий уровень интеллекта. Это связано с повышенным синтезом адреналина, поэтому такие люди излишне раздражительны, тревожны и возбуждены. Но при этом физическая нагрузка переносится плохо, сопровождается сильной мышечной и головной болью.
Смотрите на видео о синдроме Марфана:
Диагностика заболевания
Синдром Марфана
Основные критерии, по которым можно поставить диагноз (хотя бы один из них):
- расширение или расслоение аорты,
- вывих хрусталика,
- грудная клетка с выпячиванием грудины,
- конечности, длиннее, чем в норме,
- ограничение разгибания локтевых суставов или деформация бедренных.
Если пациент сгибает кисть, сжимая большой палец, то его кончик выглядывает из кулака, длина среднего пальца превышает 10 см, а соотношение длины кисти в см и роста в метрах больше 11%, большим пальцем и мизинцем можно легко охватить запястье.
Данные инструментальных методов:
- ЭКГ – аритмия, гипертрофия сердечной мышцы.
- ЭхоКГ – расширение аорты, нарушения строения клапанов, повреждения хорд, увеличенный левый желудочек, видоизмененный митральный клапан.
- Рентген – аорта в восходящей части и сердце увеличены в размерах.
- КТ и МРТ подтверждают отклонения в строении магистральных сосудов и сердечных клапанов или стенок.
- Аортография выявляет признаки аневризмы и расслоения аортальных оболочек.
Показано обследование офтальмолога с биомикроскопией тканей глазного яблока, травматолога для диагностики нарушений функции суставов и позвоночника.
Подтверждение генетических мутаций проводится при помощи анализа ДНК. Типичным признаком синдрома Марфана является значительное превышение нормы выделения хондроитинсульфата и гиалуроновой кислоты с мочой.
Лечение синдрома Марфана
Терапия зависит от наличия и степени выраженности симптомов. В основном используют курсы профилактического медикаментозного лечения бета-блокаторами, витаминами и антиоксидантами для предотвращения прогрессирования сердечно-сосудистых патологических изменений. Рекомендуется санаторно-курортное лечение – ванны, массаж, электросон, магнитотерапия на суставы.
Показания к хирургическим вмешательствам:
- недостаточность смыкания створок клапанов, значительный обратный ток крови;
- расширение аорты свыше 5 см;
- мешковидные аортальные аневризмы больших размеров или симптомы расслоения стенки.
Если пациентка с синдромом Марфана беременна, то при родах используют кесарево сечение, при наличии выраженной декомпенсации может быть показано прерывание беременности.
В послеоперационном периоде для профилактики эндокардита и тромбоэмболии назначается курс антибиотиков и антикоагулянтов.
Для нормализации зрения проводится лечение оперативным или лазерным методом, хрусталик при смещении удаляется и меняется на искусственный, подбирают линзы или очки.
Если имеются значительные деформации скелета, то используют протезирование суставов, пластику грудной клетки, стабилизацию поясничного или шейного отдела позвоночника.
Прогноз для больных
Чаще всего продолжительность жизни зависит от степени сердечной недостаточности или угрозы разрыва аневризмы аорты. Проведение оперативного лечения в оптимальные сроки помогает пациентам сохранить здоровье до 60 — 70 лет. Если нарушения затрагивают несколько систем организма, то из-за различных осложнений 90% больных умирают молодыми.
Правила жизни
Правила жизни при синдроме Марфана
Больные с синдромом Марфана должны соблюдать такие рекомендации:
- Низкий уровень физических нагрузок, запрет на соревнования, профессиональный спорт, а особенно борьбу, подводное плавание, тяжелую атлетику.
- Нельзя поднимать тяжести более 3 кг.
- Избегать вредных условий производственной деятельности, противопоказана работа, требующая нагрузку на зрение, тяжелый физический труд.
- Запрещено пребывание в условиях высокой радиации или температуры.
Для детей физкультуру можно проводить только в специальных группах в виде лечебной гимнастики.
Наследование синдрома Марфана в наши дни
Врожденный синдром Марфана передается по аутосомно–доминантному типу. Это значит, что его появление в семье подчиняется таким закономерностям:
- чаще болен один из родителей;
- частота появления у девочек и мальчиков одинаковая;
- вероятность возникновения у ребенка равна 50%;
- от здоровых родителей (при наличии больных бабушек, дедушек) обычно не передается;
- если провести анализ родословного древа, то выявляется вертикальный способ передачи синдрома в каждом поколении;
- не все члены семьи имеют одинаковые признаки болезни.
Следует учитывать, что в 20% случаев мутации гена не связаны с наследственностью, они могут возникать первично. В таких случаях играет роль возраст родителей.
Доказано, что чаще дети с этой аномалией развития появляются, если отец старше 35 лет. В следующем поколении больных при таком варианте наследования может и не быть (пропуск поколения), но половина их потомков окажется с этой патологией.
Поэтому всем будущим родителям, у которых в семьях были выявлены лица с синдромом Марфана, нужно перед планированием беременности обязательно пройти консультацию в медико-генетическом центре.
Рекомендуем прочитать статью о внезапной коронарной смерти. Из нее вы узнаете о причинах внезапной остановки сердца, факторах риска, оказании первой помощи, а также мерах профилактики.
А здесь подробнее об узелковом периартерите.
Синдром Марфана связан с дефектом генов, обеспечивающих прочность соединительной ткани. Основную опасность представляет аномальное строение сердца и аорты. Причиной смерти чаще всего бывает расслоение аневризмы аортальной стенки.
Кроме этого, особое строение костей скелета и нарушение зрения приводят к трудностям в социальной сфере. Диагностика в типичных случаях основана на внешних признаках, а для ее подтверждения используют инструментальные методы. Специфического лечения и профилактики болезни нет.
Источник
Русский Медицинский Сервер / Лечение в Италии / Центр лечения редких заболеваний в Милане / Синдром Марфана – лечение в Италии
Синдром Марфана относится к моногенным болезням соединительной ткани – группе различных по происхождению нозологических форм, которые объединяют наследственные нарушения обмена соединительной ткани. Большая часть этой патологии обусловлена нарушением ферментных систем, контролирующих синтез структурных белков из которых происходит синтез соединительной ткани. Почти все эти болезни, входящие в состав синдрома, приводят к тяжелым инвалидизирующим расстройствам.
Синдром Марфана впервые описан Вильямсом в 1876 г. Свое название заболевание получило от французского педиатра Марфана, наблюдавшего девочку с характерным симптомокомплексом болезни 20 лет спустя. Существует интересный факт, что первая девушка модель – Лесли Хорнби, которая послужила прототипом образа всех моделей, имела синдром Марфана. Так, установлено, что ряд всемирно известных людей страдали синдромом Марфана, среди них следует упомянуть президента США А. Линкольна и великого скрипача Паганини.
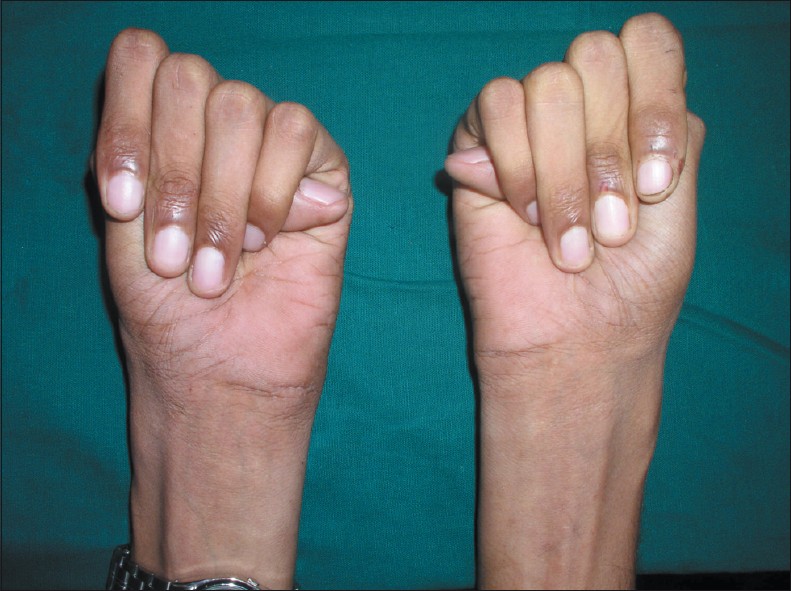
Клиническая картина заболевания характеризуется поражением многих жизненно важных органов и систем: опорно-двигательного аппарата, сердечно-сосудистой системы, органов дыхания и зрения, ЦНС.
Так, среди конституциональных особенностей и нарушений скелета наиболее часто встречаются долихопластический (астенический) тип, высокий рост (как правило, выше 180см) при выраженном дефиците массы тела (обычно ниже 50 кг.), арахнодактилия («паучьи» пальцы) кистей и стоп, кифосколиоз, воронкообразная или килевидная деформации грудной клетки, плоскостопие, узкий лицевой скелет, «готическое» небо. Не обязательно у одного человека встречаются все эти признаки, это лишь наиболее распространееные
Типичны также антимонголоидный разрез глаз, «крупный» нос, большие низкорасположенные ушные раковины, «птичье» выражение лица. У новорожденных из перечисленных особенностей скелета, как правило, выявляются только долихопластический тип и арахнодактилия. Остальные симптомы формируются в более поздние периоды развития (обычно в течение первых семи лет жизни ребенка).
Поражение сердца и сосудов – один из кардинальных признаков синдрома Марфана. Наиболее типичны среди них – пролапс митрального клапана и аневризма аорты. Сердечно-сосудистые нарушения регистрируются уже на первом-втором годах жизни ребенка, при этом отмечается постепенное увеличение диаметра аорты, достигающее критических размеров (до 6 см и более), чаще в возрасте от 16 до 45 лет.
Грозным осложнением аневризмы аорты является расслоение ее стенок, которое может быстро прогрессировать, захватывая всю длину аорты и отходящие от нее сосуды. Такие осложнения, как правило, заканчиваются летально.
Бронхолегочная система также вовлекается в патологический процесс при синдроме Марфана. Предпосылкой для этого являются механическое сдавление дыхательных путей при деформациях грудной клетки и изменения соединительнотканных структур легочной ткани. Нарушения органов дыхания в виде спонтанного пневмоторакса, легочной эмфиземы, инфаркта легкого встречаются с частотой от 10 до 75%. Наряду с этим, имеются сведения о врожденном недоразвитии одной из долей легкого, поликистозе легких, врожденной буллезной эмфиземе, двусторонних бронхоэктазах.
Диагноз синдрома Марфана основывается на семейном анамнезе, наличии у больного типичных диагностических признаков по результатам физикального осмотра, ЭКГ и ЭхоКГ, офтальмологического и рентгенологического обследования, молекулярно-генетического анализа и лабораторных исследований.
За диагностические критерии синдрома Марфана берутся характерные изменения в различных системах и органах; главными (большими) из них считаются: дилатация корня/расслоение восходящей части аорты, эктопия хрусталика и эктазия твердой мозговой оболочки; килевидная/воронкообразная деформация грудной клетки, требующая хирургического лечения; отношение длины верхнего сегмента тела к нижнему < 0,86 или размаха рук к росту > 1,05; сколиоз (> 20?) или спондилолистез; ограничение разгибания в локтевом суставе (<170о); плоскостопие; протрузия вертлужной впадины. Остальные проявления относятся к малым критериям, а генетические (семейные) признаки – к дополнительным. Для установления диагноза синдрома Марфана необходимо наличие минимум по 1-му большому критерию в двух системах органов и 1-го малого – в третьей; в скелетной системе – присутствие минимум 4-х больших.
Также применяются фенотипические диагностические тесты, определяющие соотношение кисть/рост (при синдроме Марфана > 11%); длину среднего пальца (> 10 см); индекс телосложения Варги – (масса тела, г/(рост, см)x2 – возраст, годы/100, должно быть <1,5); тест большого пальца на арахнодактилию, тест охвата запястья.
ЭКГ при синдроме Марфана позволяет определить нарушение ритма сердца, выраженную гипертрофию миокарда; ЭхоКГ – обнаружить клапанную регургитацию, увеличение размеров левого желудочка, пролапс митрального клапана, разрывы хорд, дилатацию аорты. На рентгенографии грудной клетки можно увидеть расширение корня и дуги аорты, увеличение размеров сердца; на КТ и МРТ сердца и сосудов – выявить дилатацию и аневризмы аорты.
Лечение и дальнейшее наблюдение пациентов с синдромом Марфана должно осуществляться группой специалистов: офтальмологом, кардиологом, кардиохирургом, ортопедом, генетиком, терапевтом.
Лечение больных с синдромом Марфана направлено на профилактику прогрессирования заболевания и развития осложнений, в первую очередь в сердечно-сосудистой системе. При диаметре аорты до 4 см назначаются ?-адреноблокаторы, антагонисты кальция или ингибиторы АПФ. Хирургическое лечение проводится при недостаточности клапанов сердца, пролапсе митрального клапана, значительном расширении (>5 см) восходящей части и расслоении аорты. Реконструктивные операции на аорте при синдроме Марфана, имеют высокий процент послеоперационной 5-ти и 10-ти летней выживаемости.
При необходимости выполняют протезирование митрального клапана. У беременных с синдромом Марфана и выраженной сердечно-сосудистой патологией проводят досрочное оперативное родоразрешение путем кесарева сечения. С целью профилактика инфекционного эндокардита и тромбозов после операционных вмешательств назначаются антибиотики и антикоагулянты.
При синдроме Марфана проводится коррекция зрения с помощью подбора очков и контактных линз, при необходимости – лазерное или хирургическое лечение катаракты, глаукомы, удаление смещенного хрусталика с имплантацией искусственного. При выраженных скелетных нарушениях может потребоваться хирургическая стабилизация позвоночника, торакопластика, эндопротезирование тазобедренных суставов. Применяются также патогенетическая коллагеннормализующая терапия, метаболическая и витаминотерапия.
! Несмотря на то, что многие из описанных в данном разделе болезней считаются неизлечимыми, в Центре лечения редких заболеваний в Милане постоянно ведется поиск новых методов. Благодаря генной терапии удалось добиться выдающихся результатов и полностью излечить некоторые редкие синдромы.
Обратитесь к консультанту на сайте или оставьте заявку – так вы можете узнать, какие методы предлагают итальянские врачи. Возможно, данное заболевание уже научились лечить в Милане.
+7 (925) 50 254 50 –
срочное лечение в Италии
ЗАПРОС в КЛИНИКУ
Источник