
Мукополисахаридоз 1 типа синдром гурлера фото

Мукополисахаридоз – общее название ряда редких заболеваний, которые носят генетический характер. Патология развивается вследствие нехватки в организме некоторых ферментов, помогающих расщеплять жиры и углеводы на простые молекулы. В данной статье рассмотрен мукополисахаридоз 1 типа – синдром Гурлера.
Причины
Болезнь является наследственной по аутосомно-рецессивному типу. Она развивается по причине аномалий обмена мукополисахаридов.
Патогенез
Мукополисахаридоз относится к так называемым лизосомным болезням накопления. В результате дефицита лизосомных ферментов затрудняется катаболизм гликозаминогликанов. Они накапливаются в тканях и органах, нарушая работу организма и его систем. В первую очередь происходит поражение скелета и задержка физического развития.
Внешние признаки и симптомы заболевания
Симптомы заболевания проявляются в виде дефектов костной, соединительной, хрящевой тканей. Основной признак – задержка роста. Этот симптом можно обнаружить раньше остальных, обычно уже к концу первого года жизни становится понятно, что ребенок отстает в росте.
Также мукополисахаридоз можно предположить, видя грубые черты лица. У больных большой язык, гипертелозирм (слишком большое расстояние между парными органами, в данном случае между глазами), деформированные ушные раковины, лоб нависает, зубы искривлены.

К симптомам мукополисахаридоза относятся деформации грудной клетки, выраженный кифоз грудопоясничного отдела позвоночника. При проведении рентгена можно обнаружить преждевременное окостенение затылочно-теменного шва без нарушения ядер окостенения.
В большинстве случаев болезнь сопровождает ограничение подвижности суставов, абдоминальные грыжи, гепатоспленомегалия (увеличение печени и селезенки из-за патологических процессов, происходящих в результате заболевания).
Со стороны неврологии отмечаются двигательная заторможенности и мышечная гипотония. Также при мукополисахаридозе происходит ослабление слуха и снижение интеллекта вплоть до тяжелого слабоумия. Вследствие прогрессирующих системных поражений скелета внутренние органы также подвергаются нарушениям в различной степени.
Типы мукополисахаридоза
Различают несколько типов заболевания, которые различаются выраженностью костных изменений и нарушений психики:
- I – синдром Гурлера.
- II – синдром Гунтера (Хантера).
- III – синдром Санфилиппо.
- IV – синдром Моркио.
- VI – синдром Марото – Лами.
- VII – синдром Слая.
Деление в медицинских практиках разных стран может отличаться. В тип V обычно выделяют синдром Шейе. В американском же сообществе больных мукополисаридозами делят по тяжести проявления симптоматики первого типа и выделяют три фенотипа: синдром Гурлера, синдром Шейе и промежуточный между ними синдром Гурлер – Шейе (из них Гурлер самый тяжелый, Шейе – самый легкий).
Синдром Гурлера
Эта форма встречается чаще остальных и была описана ранее других синдромов. К тому же клиническая картина наиболее яркая и типичная из всех видов мукополисахаридоза.
Синдром Гурлера развивается в результате аутосомно-рецессивного наследования. Этот тип болезни характеризуется очень быстрым прогрессом. Несмотря на то что мукополисахаридоз первого типа схож со вторым (Гунтера, или Хантера), это более сложное заболевание. Впервые данная форма была описана в 1919 году Гертрудой Гурлер (поэтому правильное название – синдром Гурлер, а не Гурлера). Частота появления – один случай на 20-25 тысяч человек, причем в большинстве случаев родители заболевшего ребенка находятся в кровном родстве. Поэтому если ставится диагноз “синдром Гурлера”, причины необходимо искать на генетическом уровне. Симптомы проявляются практически сразу после рождения, а к году-двум клиническая картина уже выражена полностью.
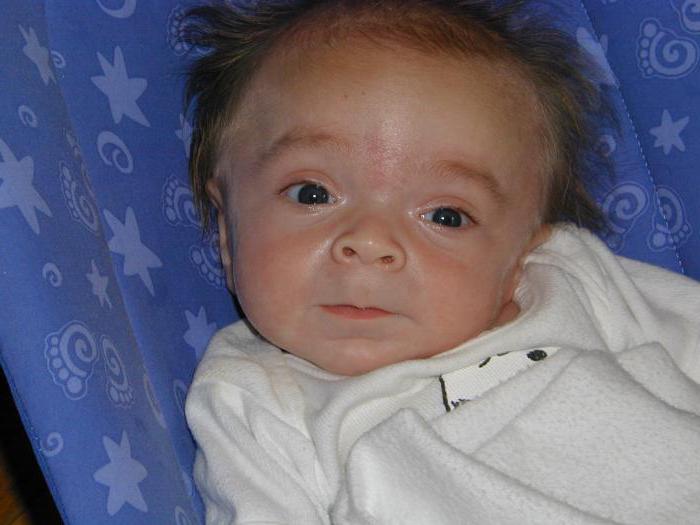
Синдром Гурлера – классическое проявление заболевания. По мере развития недуга замедляется рост, отмечается видимое помутнение роговицы, сосуды носа переполняются кровью. При этой форме заболевания при рентгенологическом исследовании можно обнаружить расширение турецкого седла, укорочение и расширение длинных костей, гипоплазию и заостренность позвонков поясничной области (так называемые рыбьи позвонки), деформации позвоночного столба (больные страдают кифозом и лордозом грудопоясничной области позвоночника). Начинаются патологии сердечно-сосудистой системы – закупориваются коронарные артерии, изменяются клапаны, миокард, эндокард, сердце увеличивается в размерах.
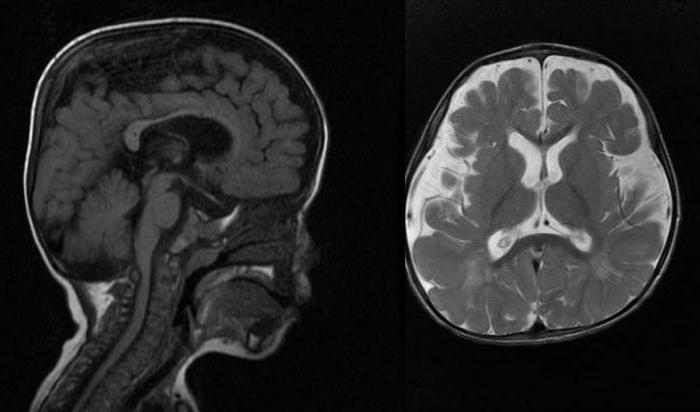
Отмечается гидроцефалия, причиной которой становятся отложения мукополисахаридов в мозговых оболочках. Определяются очаги демиелинизации. Мукополисахариды также откладываются в печени, селезенке, эпителии почечных канальцев; сетчатке, склере, роговице глаз; нервных клетках, хрящах.
Дети рождаются уже с характерным внешним видом – у них очень своеобразные, грубые черты лица, из-за чего другое название мукополисахароидозов – гаргоилизм (от слова «гаргулья» – фантастическая фигура с необычными чертами лица), в том числе так называют и синдром Гурлера. Фото, на которых изображены больные, наглядно иллюстрируют причудливое искажение черт лица ребенка. У таких детей изменен череп – он приобретает форму киля лодки, наблюдается так называемая скафоцефалия, запавшая переносица, толстые губы, большой язык, крутой лоб, короткая шея и характерное выражение лица. Внешне это действительно напоминает то, как изображают мифологических гаргулий.

Также у них укорочена грудная клетка, нижние ребра выступают, есть признаки кифоза, суставы (особенно пальцев и локтей) малоподвижны, могут быть паховые и пупочные грыжи. Ногти могут приобретать вид часовых стекол, волосы становятся жесткими и сухими, голос – низким и хриплым. Вероятна тугоухость или даже глухота. Больные часто страдают от кариеса, который провоцирует синдром Гурлера.
Симптомы включают патологии дыхательной системы, из-за этого ребенок дышит ртом, у него появляются аденоиды, он подвержен вирусным инфекциям. Со временем у него развиваются характерные для мукополисахаридозов проблемы с печенью и селезенкой (в результате увеличен живот), слабоумие.
Рост остается карликовым. Из-за неправильного телосложения и деформации позвоночника больные ходят на полусогнутых ногах, на цыпочках.
У синдрома Гурлера злокачественный прогрессирующий характер, поэтому крайне быстро наступает инвалидизация больных. Многие не доживают даже до 10 лет.
Диагностика
Пациенту необходимо провести клиническое, рентгенологическое, биохимическое, генеалогическое, а также молекулярно-генетические исследования. Диагностика проводится по клиническим проявлениям болезни, на основе рентгенологических исследований и анализа мочи, по которому определяется активность ферментов и экскреция гликозаминогликанов.
Лечение мукополисахаридоза
Если больному поставлен диагноз “синдром Гурлера”, лечение предполагается больше симптоматическое. Пациент наблюдается комплексно у ортопеда, хирурга, педиатра, отоларинголога, нейрохирурга, офтальмолога и невропатолога. Больному проводится ортопедическая коррекция нарушений опорно-двигательного аппарата, удаляют грыжи, лечатся часто возникающие у таких пациентов вирусные заболевания, нарушения слуха, отиты, синуситы. Также под наблюдение попадает сердечно-сосудистая система.
Используются гормональные препараты, которые временно улучшают состояние пациента:
- глюкокортикоиды,
- кортикотропин,
- тиреоидин.
Помимо этого больному показаны витамин А, декстран 70, которые также временно улучшают состояние больного. Недолгосрочное улучшение дает переливание препаратов плазмы крови.
При синдроме Гурлера пациенту может быть назначено физиотерапевтическое лечение: электрофорез лидазы на область пораженных суставов, лазерная пунктура, магнитотерапия, парафиновые аппликации. Также больным рекомендуется заниматься лечебной физкультурой, упражнения которой воздействуют на суставы и позвоночник. Хорошие результаты часто дает массаж.
Так как больные синдромом Гурлера подвержены респираторным заболеваниям, необходимо своевременно проводить санацию очагов инфекций во рту и носоглотке.
Для лечения мукополисахаридоза 1-го типа часто проводятся хирургические вмешательства – пересадка роговицы и коррекция клапанных пороков сердца и ущемлений нервов. В международной практике помимо симптоматического лечения медикаментами, физиотерапией или хирургических вмешательств используется заместительная ферментная терапия, а также трансплантация стволовых клеток.
При необходимости больному проводят грыжеиссечения, удаление аденоидов, антиглаукоматозные операции, трахеостомию, протезирование тазобедренных суставов, шунтирование при гидроцефалии и пр.
Прогноз
Прогноз неблагоприятен как для синдрома Гурлера, так и для других форм, которые имеет мукополисахаридоз. Синдром Гурлера отличается наибольшей безнадежностью. Изменения скелета с каждым годом увеличиваются, в результате органы и системы подвергаются более значительным нарушениям. Если ребенок не умирает от пневмонии в раннем возрасте, то к 7-12 годам это уже беспомощный физически и психически инвалид. До юношеского возраста доживают единицы.
Профилактика
Предупредить данное заболевание невозможно. Но можно обнаружить его на самой ранней стадии – при пренатальной диагностике. Для этого проводится анализ амниотических клеток на предмет дефицита фермента (в случае положительного результата беременной рекомендуется аборт).
За счет ранней диагностики и своевременной терапии развившейся компрессии спинного мозга можно избежать необратимых повреждений нервов. Для профилактики обязательно проводится медико-генетическая консультация.
Прогнозы лечения
Несмотря на сложности в плане лечения синдрома Гурлера, в последние 20 лет во многих развитых странах используется трансплантация костного мозга, которая значительно улучшает качество жизни больных. Более 10 лет применяются препараты заместительной терапии для лечения всех проявлений мукополисахаридоза не неврологического характера.
Источник
Содержание
В этом году исполняется ровно 100 лет с тех пор, как канадец Чарльз Хантер составил первое описание мукополисахаридоза, выявленного у двух братьев. Сегодня известно, что это не одно, а целая группа заболеваний, очень редких. В мире проживает примерно 1,5 тысячи таких пациентов, около 200 из них живут в России. MedAboutMe выяснял, что такое мукополисахаридоз и какова судьбы людей, больных этим заболеванием.
Мукополисахаридоз: ферменты и болезнь
Немалая часть нашего организма состоит из соединительной ткани. Ее клетки участвуют в формировании каркаса и наружного покрова всех органов тела. В некоторых их них соединительная ткань составляет до 90% от их массы. Это хрящи, кости, жир, фасции, синовиальная жидкость, которая плещется в суставах, лимфа, склера и радужка глаза, микроглия, окружающая нейроны и др.
Без преувеличения, главным элементом соединительной ткани можно считать протеогликаны. Это соединения, которые содержат белковое ядро и связанные с ним многочисленные и разнообразные гликозаминогликаны (ГАГ). В разных тканях — разные протеогликаны. Они образуются, живут 7-10 дней и распадаются под действием ферментов — лизосомных гидролаз. Последние расщепляют именно ГАГ, причем каждому виду гликозаминогликанов соответствует свой личный фермент. Процесс образования (анаболизма) протеогликанов и распада (катаболизма) идет постоянно.
Что произойдет, если нужного фермента не окажется? Нерасщепленные или частично разрушенные протеогликаны станут накапливаться, откладываясь в соединительных тканях, пронизывающих все наше тело. Так и развиваются мупоколисахаридозы — разновидность лизосомных болезней накопления.
Какие бывают мукополисахаридозы?
На сегодняшний день известно о 8 типах заболевания, причем некоторые из них представлены в нескольких разновидностях. Обычно это связано с тем, что дефицит одного фермента может быть вызван разными мутациями — и, как следствие, проявления этих мутаций и срок жизни больных тоже могут различаться.
- I тип объединяет три фенотипа: синдром Гурлер (МПС-IH), синдром Шейе (МПС-IS) и синдром Гурлер-Шейе (МПС-IH/S). Мутации, вызывающие синдром Гурлер и синдром Шейе, происходят в одном и том же гене. И иногда они развиваются одновременно. МПС I типа считается самой распространенной разновидностью болезни. Продолжительность жизни — от 6-10 (синдром Гурлер) до 30 лет (синдром Шейе).
- II тип — синдром Хантера (МПС-II). Это второй по частоте встречаемости вид мукополисахаридозов. Продолжительность жизни — 30-40 лет.
- III тип — синдром Санфилиппо, который подразделяется на 4 формы: A (МПС-III A), B (МПС-III B), C (МПС-III C) и D (МПС-III D). Это тоже разные мутации гена, который кодирует данный лизосомный фермент. Продолжительность жизни — до 20 лет.
- IV тип — синдром Моркио. Представлен двумя формами: A (МПС-IV A) и B (МПС-IV B). Продолжительность жизни — 20-35 лет.
- VI тип — синдром Марото-Лами (МПС-VI). Продолжительность жизни — 10-20 лет.
- VII тип — синдром Слая (МПС-VII).
- VIII тип — синдром Ди Ферранте (МПС-VIII) .
- XI тип — синдром дефицита гиалуронидазы, он же — синдром Натовича (МПС-IX).
Пятым типом раньше считался синдром Шейе.
Диагноз обычно ставят в детском возрасте.
МПС и генетика
Мукополисахаридоз — это генетическое заболевание. Оно наследуется по аутосомно-рецессивному типу. Это значит, что мутация только на одной хромосомы из пары (а мы помним, что у человека 23 пары хромосом) не вызовет заболевания, потому что ген, расположенный на парной нормальной хромосоме будет исправно работать. Благодаря этому организм будет производить нужный фермент в требуемых количествах. А вот если человек получит на обеих хромосомах одинаковые мутации в области генов, кодирующих один и тот же лизосомный фермент — у него разовьется мукополисахаридоз. Чтобы такая ситуация сложилась, оба его родителя должны быть носителями бракованного гена. Такое совпадение случается очень редко — и поэтому мукополисахаридозы относятся к орфанным (редким) заболеваниям.
Из общего списка выбивается синдром Хантера (или Гунтера, в зависимости от перевода), МПС II типа. Именно это заболевание стало первым из открытых человечеством мукополисахаридозов. В отличие от генов, являющихся причиной других видов МПС, ген, вызывающий синдром Хантера, расположен на половой X-хромосоме. То есть его наследование сцеплено с полом, и болеют им в подавляющем большинстве случаев мальчики.
- При других видах МПС, если болен один родитель, а второй здоров, ребенок получит одну хромосому с мутантным геном, а другую — такую же, но нормальную. Болезнь не разовьется.
- Но половые хромосомы Х и Y отличаются друг от друга. Мальчики получают Y-хромосому от отца и X-хромосому от матери. Если мать была обладателем хотя бы одной Х-хромосомы с мутантным геном (то есть даже сама не болела, была лишь носителем), и эта «неправильная» хромосома вошла в геном ребенка, то второй здоровой хромосомы ей в пару нет. Такой мальчик заболевает.
«Люди-горгульи» и другие проявления болезни
Разные типы мукополисахаридоза имеют свои характерные особенности. Например, при МПС I, II и III типов развиваются тяжелые нарушения нервной системы, которые приводят к деменции (слабоумию). А есть и общие черты. Как мы помним, речь идет о заболеваниях, связанных с соединительной тканью. Поэтому неудивительно, что на их фоне развиваются тяжелые системные поражения костей, суставов, кожи.
При этом может меняться форма головы, а при некоторых типах МПС уже с детства формируется специфическая внешность: приплюснутая переносица и нос с крупными ноздрями, широко посаженные глаза, толстые губы и приоткрытый рот, в целом грубые выразительные черты лица. Подметив сходство лиц таких пациентов с горгульями — каменными изображениями мифических существ характерного облика, врачи дали название данному состоянию — гаргоилизм (от французского слова gargouille).
Сегодня выделяют два основных фенотипа (внешние проявления) мукополисахаридоза:
- Гурлер-подобный фенотип. В этом случае обычно развиваются деменция, гепатоспленомегалия (увеличение печени и селезенки) и гаргоилизм.
- Моркио-подобный фенотип. У таких пациентов нормальный интеллект и не настолько выражены повреждения скелета и связок, как при Гурлер-подобном фенотипе.
Все пациенты с МПС также страдают от задержки роста (нанизм) и диспропорционального развития скелета, нарушений зрения (помутнение роговицы и др.), слуха (тугоухость), заболеваний сердца и сосудов (аритмии, гипертрофия миокарда, заболевания клапанов сердца), болезней дыхательной системы.
Лечение мукополисахаридозов
Специфическая терапия
Специфической называется терапия, мишенью которой являются непосредственные причины болезни. В данном случае это ферменты, которые не вырабатываются в достаточном количестве, и те нерасщепленные мукополисахариды, которые накапливаются и вызывают болезнь.
Только в середине прошлого века стали появляться препараты, позволяющие заменить недостающие ферменты. Сегодня производятся — и зарегистрированы в России — синтетические ферменты для трех типов МПС:
- МПС-I — Альдуразим (ларонидаза);
- МПС-II — Элапраза (идурсульфаза);
- МПС-VI — Наглазим (галсульфаза).
Недостаток Элапразы заключается в том, что она не проникает сквозь гематоэнцефалический барьер, то есть не попадает в мозг. В 2015 году СМИ сообщали о начале лечения 31-летнего пациента с синдромом Хантера новым препаратом AGT-182 — комбинацией идурсульфазы и специально разработанного для этого антитела. Предполагалось, что это позволит лекарству проникнуть в мозг. Результаты эксперимента пока не опубликованы.
Специфических лекарств для лечения других типов МПС в России пока нет. Только симптоматическая терапия, направленная на уменьшение проявлений болезни, дает возможность таким больным прожить отведенный им срок.
В 2014 году FDA одобрило препарат Вимизим (элосульфаза альфа), предназначенный для лечения МПС-IV А — разновидности синдрома Моркио. В России данный препарат не зарегистрирован.
Другой путь — субстратредуцирующая терапия, то есть угнетение выработки мукополисахаридов, избыток которых приводит к болезни. Пока такое лекарство разработано для МПС I, II и III типов (генистеин), но исследования проводились только на животных. В июне этого года ученые из Университета Манчестера сообщили о запуске проекта, в котором участвуют дети, больные МПС. Проект будет длиться год, и исследователи рассчитывают, что им удастся остановить разрушение центральной нервной системы при этом заболевании.
Трансплантация костного мозга
Ферментозаместительная терапия основана на введении экзогенных (то есть извне) синтетических ферментов. А трансплантация костного мозга (ТКМ) призвана заставить организм вырабатывать собственные, эндогенные ферменты при помощи пересаженных донорских стволовых клеток.
ТКМ — крайне сложная и очень дорогостоящая процедура. Она подходит далеко не всем пациентам и не со всеми типами МПС. Лучшие результаты были получены при пересадке костного мозга детям, страдающим МПС I типа, в возрасте 2 лет, то есть до того момента, пока поражение центральной нервной системы не стало слишком обширным. При попытках лечения методом ТКМ мукополисахаридозов II типа отмечается повышенная частота осложнений и летальность среди пациентов. Следует учитывать, что и сам процесс подбора неродственного донора возможен далеко не для всех пациентов.
Генная терапия
Одним из самых значимых направлений в области разработки методов лечения МПС является генная терапия при помощи вирусных векторов — кольцевых ДНК, способных переносить нужный ген и встраивать его в заданный участок хромосомы. И уже есть первые положительные результаты.
В июле этого года ученые из Университета Миннесоты рассказали, как закапали в нос экспериментальным мышам препарат, содержащий вирус с геном, кодирующим лизосомный фермент альфа-L-идуронидазу — именно его так не хватает больным с МПС I типа. Введение препарата через нос позволило ему проникнуть через гематоэнцефалический барьер в мозг и защитить мозг от разрушительного действия накапливающихся мукополисахаридов. Теперь ученые ищут возможности повторить ту же самую процедуру с участием людей.
Мукополисахаридоз в России
С 2012 года мукополисахаридоз входит в список жизнеугрожающих орфанных заболеваний и подлежит лечению по программе «7 нозологий». Если быть точными, то в список вошли только те типы МПС, от которых есть лекарства и при этом они зарегистрированы в нашей стране, то есть: МПС-I, МПС-II и МПС-VI.
Сегодня в России, по данным общественных организаций, занимающихся проблемой МПС, проживает около 200 пациентов с мукополисахаридозом. А по данным компании Санофи, в 2017 году диагноз МПС имели 98 россиян, причем 88 из них — это дети. Сегодня лечение получают только 77 из них: 52 человека находятся на пожизненной ферментозаместительной терапии, а 25 — прошли трансплантацию костного мозга (при синдроме Гурлер эта операция может проводиться только в возрасте от 2 до 4 лет). Из-за высокой стоимости лекарств, которые следует вводить еженедельно, расходы на это заболевание лидируют в бюджетах, выделенных на лечение орфанных болезней.
Например, для лечения людей с синдромом Хантера необходим препарат Элапраза. Стоимость одного флакона колеблется от 100 до 200 тысяч рублей. Для лечения одного пациента еженедельно требуется 300-500 тысяч рублей. И это пожизненная терапия. Должна быть.
В последние годы закупкой препаратов для пациентов с МПС занимались региональные власти. Нежелание тратить огромные деньги на отдельных пациентов с редкими болезнями приводило к бесконечным задержкам поставки лекарств — а это немедленно ухудшало состояние больных. Но недавно, в июне стало известно, что финансирование закупок препаратов для лечения МПС с будущего, 2018 года будет производиться из федерального бюджета. Это дает надежду многим из нынешних пациентов дожить до того момента, когда их болезнь можно будет вылечить.
- Мукополисахаридоз I типа: новая технология лечения – ферментозамещающая терапия / Семячкина А.Н., Новиков П.В., Воскобоева Е.Ю., Яблонская М.И., Курбатов М.Б., Харабадзе М.Н. // Российский вестник перинатологии и педиатрии. = 2012. – Т. 57 №4-1. – с. 94-102
- Скелетные проявления при мукополисахаридозах различных типов / Бучинская Н.В., Костик М.М., Чикова И.А., Исупова Е.А., Калашникова О.В., Часнык В.Г., Губин А.В., Рябых С.О., Очирова П.В. // Гений ортопедии. = 2014. – 2. – с. 81-90
Источник