
Мукополисахаридоз код мкб 10

Утратил силу — Архив
РЦРЗ (Республиканский центр развития здравоохранения МЗ РК)
Версия: Архив – Клинические протоколы МЗ РК – 2010 (Приказ №239)
Категории МКБ:
Мукополисахаридоз, тип I (E76.0)
Общая информация
Краткое описание
Мукополисахаридозы являются следствием генерализованного процесса, который поражает главным образом соединительную ткань в результате метаболического дефекта, приводящего к накоплению определенных веществ, как в соединительной ткани, так и в мозге.
Мукополисахаридозы – это класс заболеваний, относится к обширной группе нарушений обмена гликозаминогликанов. К настоящему времени известно, по крайней мере, 11 нарушений обмена мукополисахаридов с различным первичным биохимическим дефектом, т.е. дефицитом разных ферментов. Вся группа объединена повышенным накоплением в клетках кислых мукополисахаридов и повышенной экскрецией этих веществ с мочой. Большинство из этих заболеваний характеризуется изменениями скелета и внутренних органов, выраженными при разных формах в различной степени, сопровождается грубыми нарушениями нервной системы, приводящими к тяжелому слабоумию.
Протокол “Мукополисахаридозы тип I; тип II”
Код по МКБ-10: Е76,0; Е76,1
Синдромы:
– Гурлер
– Гурлер-Шейе
Облачная МИС “МедЭлемент”
Облачная МИС “МедЭлемент”
Классификация
1. Мукополисахаридоз типа I (синдром Гурлер).
2. Мукополисахаридоз типа II (синдром Гунтера).
3. Мукополисахаридоз типа III (синдром Санфилиппо).
4. Мукополисахаридоз типа IV (синдром Моркио).
5. Мукополисахаридоз типа V (Шейе).
6. Мукополисахаридоз типа VI (синдром Марото-Лами).
Диагностика
Диагностические критерии
Жалобы и анамнез: задержка в психоречевом развитии, снижение мышления, памяти, внимания, расторможенность, искривление позвоночника, низкий рост, грыжи, снижение зрения, увеличение размеров головы, грубые черты лица. Дети рождаются без внешних изменений, в первые месяцы жизни или позже черты лица становятся грубыми, дети отстают в психо-речевом развитии, росте.
Физикальное обследование: больные разными формами мукополисахаридозов фенотипически очень похожи друг на друга: у них отмечаются гротескные черты лица, деформации скелета, изменения внутренних органов (пороки сердца, гепато- и спленомегалии), пупочные и паховые грыжи, снижение интеллекта, помутнение роговицы, карликовый рост.
Синдром Гурлера. Дети рождаются без внешних изменений, в первые месяцы жизни черты лица становятся грубыми, характерны запавшая переносица, помутнение роговицы, гепатоспленомегалия, тугоподвижность голеностопных суставов, кифоз. Постоянными симптомами, развивающимися на втором году жизни, являются короткая шея, воронкообразная или килевидная грудная клетка, паховые и пупочные грыжи, гипертрихоз, макро- и скафоцефалия, увеличение языка и губ, мелкие, редко посаженные зубы, шумное дыхание, ограничение подвижности в межфаланговых, локтевых, плечевых и тазобедренных суставах. Кожа сухая, грубая, бледная. Позже появляются признаки поражения сердца: шум, кардиомегалия. Развиваются глухота, слепота. Рост замедляется после первого года жизни. Психомоторное развитие протекает нормально до 2 лет. Для поздних стадий заболевания характерна глубокая деменция.
Синдром Гунтера. Данный тип мукополисахародоза характеризуется доброкачественным течением. До 1-2 лет выраженных клинических проявлений не наблюдается, за исключением шумного дыхания вследствие обструкции верхних дыхательных путей, повторных ринитов, макро- и скафоцефалия, паховых и пупочных грыж. В возрасте старше 2 лет появляются утолщение ноздрей, губ и языка, тугоподвижность суставов, задержка роста, гипертрихоз и гепатоспленомегалия. Частыми признаками болезни являются утолщенная кожа, короткая шея, редкие зубы. Интеллект сохранен, но могут наблюдаться изменения психики.
Лабораторные исследования:
– общий анализ крови – с целью исключения анемии;
– общий анализ мочи – с целью исключения уронефрологической патологии.
Инструментальные исследования:
1. Электромиография (ЭМГ) выявляет снижение амплитуды двигательных потенциалов, спонтанная двигательная активность не характерна.
2. Компьютерная томография головного мозга (КТ) – картина гипертензионно-гидроцефального синдрома.
3. Электроэнцефалография (ЭЭГ) выявляет нарушение электрической активности – плохая выраженность альфа-ритма в затылочной области, наличие медленных волн в передних отделах мозга.
4. ЭКГ.
5. ЭХО кардиография.
6. УЗИ органов брюшной полости, почек.
7. Аудиограмма.
8. Осмотр глазного дна.
Показания для консультации специалистов:
– логопед;
– психолог;
– ЛОР сурдолог;
– окулист;
– кардиолог;
– генетик;
– педиатр;
– ортопед;
– протезист;
– врач ЛФК;
– физиотерапевт.
Минимум обследования при направлении в стационар:
1. Общий анализ крови и мочи.
2. АЛТ.
3. АСТ.
4. Кал на яйца глист.
Основные диагностические мероприятия:
1. Общий анализ крови.
2. Общий анализ мочи.
3. ЭЭГ – для выявления эпилептической активности.
4. Исследование слуха – уточнение остроты слуха, исключения тугоухости.
5. КТ головного мозга – с целью исключения гидроцефалии, врожденных пороков развития головного мозга.
6. Логопед – уточнение речевых нарушений, назначение коррекционных занятий.
7. Психолог – исследование психического развития.
8. Окулист – осмотр глазного дна, выявление глазной патологии.
9. Ортопед – уточнение ортопедической патологии.
10. Генетик.
11. ЭКГ.
12. ЭХО кардиография.
13. Кардиолог.
14. УЗИ органов брюшной полости.
15. Педиатр.
16. Хирург-уролог.
Дополнительные диагностические мероприятия:
1. ИФА на токсоплазмоз.
2. ИФА на цитомегаловирус.
3. Анализ мочи на обменные нарушения.
Дифференциальный диагноз
Признак Синдром | Начало | Минимальные диагностические признаки | Соматические проявления | Лабораторные данные |
Синдром Гурлера | На первом году жизни проявляется увеличением и деформацией черепа, задержкой физического и умственного развития | Грубые черты лица, задержка роста, выраженная умственная отсталость, короткая шея, воронкообразная или килевидная грудная клетка, искривление позвоночника, паховая и пупочные грыжи, ограничение подвижностей в суставах | Поражение сердца, часто воспалительные заболевания верхних дыхательных путей, пневмонии, гепатоспленомегалия | Повышенная экскреция с мочой мукополисахаридов – дерматансульфата и гепарансультата |
Синдром Гунтера | 1-2 года | Макро- и скафоцефалия, утолщение ноздрей, губ и языка, тугоподвижность суставов, задержка роста, гипертрихоз, утолщенная кожа, короткая шея, редкие зубы | Гепатоспленомегалия, изменения клапанного аппарата сердца | Мукополисахоридурия, дефицит идуронат-сульфатазы в фибробластах, сыворотки крови, лимфоцитах |
Синдром Санфилиппо | В возрасте 1-4 года проявляется повышенной возбудимостью, нарушением внимания, агрессивностью | Гипертензионно-гидроцефальный синдром, ретинопатия, прогрессирующее снижение интеллекта, нарушение речи, спастическая диплегия. Характерно: выступающий лоб, гипертелоризм, толстые губы, большой язык, низкий рост, короткие, широкие конечности | Гепатоспленомегалия | Повышенная экскреция с мочой гепаратсульфата, кислых гликозаминогликанов |
Синдром Моркио | На 2 году жизни – отставание в росте и разнообразные деформации скелета | Отставание в росте, скафоцефалия, кифосколиоз, прогрессирующие деформации позвоночника и грудины, короткая шея, килевидная грудная клетка, поясничный лордоз | Пороки сердца, большой живот | В моче – избыток кератансульфата или всех кислых гликозаминогликанов |
Синдром Шейе | Школьный возраст | Характерно широкое лицо, отставание в росте, умеренная мышечная гипотония, короткая шея, широкие и короткие кисти и стопы, снижение зрения и слуха | Аномалия аортального клапана, иногда гепатоспленомегалия | Повышена экскреция с мочой кератан- и дерматансульфата |
Синдром Марото-Лами | С 2-3 лет отмечается отставание в росте, прогрессирующее помутнение роговицы | Умеренные проявления внутричерепной гипертензии, к школьному возрасту появляются грубые черты лица, помутнение роговицы, короткая шея, бочкообразная грудная клетка, синдром запястного канала, поясничный кифоз, грыжи | Гепатоспленомегалия, сердечнососудистая и дыхательная недостаточность | Дефицит арилсульфатазы |
Онлайн-консультация врача
Посоветоваться с опытным специалистом, не выходя из дома!
Консультация по вопросам здоровья от 2500 тг / 430 руб
Интерпретация результатов анализов, исследований
Второе мнение относительно диагноза, лечения
Выбрать врача
Лечение
Тактика лечения: специфического лечения при мукополисахаридозах нет. Лечение симптоматическое.
Цели лечения:
– активизация психического развития;
– пополнение пассивного и активного словарного запаса;
– коррекция поведения;
– повышение эмоционального тонуса, настроения ребенка;
– обучение навыкам самообслуживания;
– социальная адаптация.
Немедикаментозное лечение:
– индивидуальные занятия с логопедом;
– занятия с психологом;
– кондуктивная педагогика;
– ЛФК;
– массаж;
– физиолечение – кислородный коктейль, озокеритовые аппликации, дарсонвализация волосистой части головы, электрофорез с эуфиллином на шейный отдел позвоночника.
Медикаментозное лечение
Широко используют в последнее время препараты ноотропного ряда – нейропротекторы, с целью улучшения обменных процессов в головном мозгу. Большинство ноотропных препаратов в связи с их психостимулирующим действием назначают в первую половину дня. Продолжительность курсов лечения ноотропами составляет от одного до двух-трех месяцев.
Церебролизин, ампулы 1 мл в/м, пирацетам, ампулы 5 мл 20%, гинкго-билоба (танакан), таблетки 40 мг, пиритинол гидрохлорид (энцефабол), драже 100 мг, суспензия – 5 мл содержит 80,5 мг пиритинола (соотв. 100 мг пиритинола гидрохлорида). Энцефабол – минимум противопоказаний, разрешен к применению с первого года жизни. Дозирование суспензии (с содержанием в 1 мл 20 мг энцефабола) детям 3-5 лет суточная доза 200-300 мг (12-15 мг массы тела) назначают в 2 приема – утром (после завтрака) и днем (после дневного сна и полдника). Продолжительность курса 6-12 недель, целесообразен длительный прием, при котором повышается работоспособность и способность к обучению, улучшаются высшие психические функции: актовегин, ампулы 2 мл 80 мг, драже-форте 200 мг активного вещества.
Актовегин нейрометаболический препарат, содержащий исключительно физиологические компоненты. Детям назначается в драже-форте, прием до еды по ½ -1 драже 2-3 раза в день (в зависимости от возраста и выраженности симптомов заболевания), до 17 часов. Продолжительность терапии 1-2 месяца. Инстенон, таблетки (1 таблетка содержит 50 мг этамивана, 20 мг гексобендина, 60 мг этофиллина). Многокомпонентный нейрометаболический препарат. Суточная доза составляет 1,5-2 таблетки, назначается в 2 приема (утром и днем) после еды. Для исключения побочных эффектов рекомендуется постепенное наращивание дозы в течение 5-8 дней. Продолжительность лечения 4-6 недель.
Ангиопротекторы с целью улучшения мозгового кровообращения: винпоцетин, циннаризин.
Витамины группы В: В1, В6, В12, нейромультивит – специальный комплекс витаминов группы В с направленным нейротропным действием, неуробекс, фолиевая кислота, аевит.
Седативная терапия по показаниям: ноофен, ново-пассит.
Корректоры поведения: сонапакс, хлорпротиксен.
Профилактические мероприятия:
– профилактика травматизма;
– профилактика вирусных и бактериальных инфекций.
Дальнейшее ведение: регулярные занятия с логопедом, дефектологом, психологом, социальная адаптация ребенка, оформление в специализированный детский сад, прохождение медико-педагогической комиссии для решения вопроса об обучении ребенка.
Основные медикаменты:
1. Актовегин, ампулы 2 мл 80 мг
2. Винпоцетин (кавинтон), таблетки 5 мг
3. Пирацетам, ампулы 5 мл, 20%
4. Пирацетам, таблетки 0,2 и 0,4
5. Пиридоксин гидрохлорид, ампулы, 1 мл, 5%
6. Тиамин хлорид, ампулы 5%, 1 мл
7. Фолиевая кислота, таблетки 0,001
8. Церебролизин, ампулы 1 мл
9. Цианокобаламин, ампулы 1 мл, 200 мкг и 500 мкг
Дополнительные медикаменты:
1. Аевит, капсулы
2. Глицин, таблетки 0,1
3. Гопантеновая кислота (пантокальцин), таблетки 0,25
4. Инстенон, таблетки
5. Нейромультивит, таблетки
6. Неуробекс, таблетки
7. Ново-пассит, таблетки, раствор
8. Ноофен, таблетки 0,25
9. Пиритинол (энцефабол), драже 0,1; суспензия
10. Сонапакс, таблетки 10 мг
11. Хлорпритиксен 15 мг
12. Циннаризин (стугерон), таблетки 25 мг
Индикаторы эффективности лечения:
– улучшение внимания, памяти, работоспособности;
– пополнение пассивного и активного запаса слов;
– повышение эмоционального и психического тонуса.
Госпитализация
Показания к госпитализации (плановая): задержка психоречевого и моторного развития, различные врожденные пороки развития конечностей.
Информация
Источники и литература
- Протоколы диагностики и лечения заболеваний МЗ РК (Приказ №239 от 07.04.2010)
- Е.И. Гусев, Г.С. Бурд, А.С. Никифоров. Неврологические симптомы, синдромы, симптомокомплексы и болезни. Москва 1999
Л.В. Калинина, Е.И. Гусев. Наследственные болезни метаболизма и факоматозы. Москва 1981
Л.О. Бадалян. Детская неврология. Москва 1998
Г.С. Маринчева, В.И. Гаврилов. Умственная отсталость при наследственных болезнях. Москва 1988
С.К. Козлова, Е. Семанова. Наследственные синдромы и медико-генетическое консультирование. Ленинград 1987
- Е.И. Гусев, Г.С. Бурд, А.С. Никифоров. Неврологические симптомы, синдромы, симптомокомплексы и болезни. Москва 1999
Информация
Список разработчиков:
№ | Разработчик | Место работы | Должность |
1. | Кадыржанова Галия Баекеновна | РДКБ «Аксай», психоневрологическое отделение №3 | Заведующая отделением |
2. | Серова Татьяна Константиновна | РДКБ «Аксай», психоневрологическое отделение №1 | Заведующая отделением |
3. | Мухамбетова Гульнара Амерзаевна | Кафедра нервных болезней, КазНМУ | Ассистент, кандидат медицинских наук |
4. | Балбаева Айым Сергазиевна | РДКБ «Аксай», психоневрологическое отделение №3 | Врач-невропатолог |
Прикреплённые файлы
Мобильное приложение “MedElement”
- Профессиональные медицинские справочники. Стандарты лечения
- Коммуникация с пациентами: онлайн-консультация, отзывы, запись на приём
Скачать приложение для ANDROID / для iOS
Мобильное приложение “MedElement”
- Профессиональные медицинские справочники
- Коммуникация с пациентами: онлайн-консультация, отзывы, запись на приём
Скачать приложение для ANDROID / для iOS
Внимание!
Если вы не являетесь медицинским специалистом:
- Занимаясь самолечением, вы можете нанести непоправимый вред своему здоровью.
- Информация, размещенная на сайте MedElement и в мобильных приложениях “MedElement (МедЭлемент)”, “Lekar Pro”,
“Dariger Pro”, “Заболевания: справочник терапевта”, не может и не должна заменять очную консультацию врача.
Обязательно
обращайтесь в медицинские учреждения при наличии каких-либо заболеваний или беспокоящих вас симптомов.
- Выбор лекарственных средств и их дозировки, должен быть оговорен со специалистом. Только врач может
назначить
нужное лекарство и его дозировку с учетом заболевания и состояния организма больного.
- Сайт MedElement и мобильные приложения “MedElement (МедЭлемент)”, “Lekar Pro”,
“Dariger Pro”, “Заболевания: справочник терапевта” являются исключительно информационно-справочными ресурсами.
Информация, размещенная на данном
сайте, не должна использоваться для самовольного изменения предписаний врача.
- Редакция MedElement не несет ответственности за какой-либо ущерб здоровью или материальный ущерб, возникший
в
результате использования данного сайта.
Источник
Связанные заболевания и их лечение
Описания заболеваний
Национальные рекомендации по лечению
Стандарты мед. помощи
Содержание
- Описание
- Симптомы
- Лечение
- Прогноз
- Профилактика
- Основные медицинские услуги
- Клиники для лечения
Названия
Мукополисахаридоз.
Дефицит ферментов при мукополисахаридозах
Описание
Мукополисахаридозы – это группа наследственных заболеваний, вызванных неполным разрушением и накоплением кислых мукополисахаридов (гликозоаминогликанов).
Клиника обусловлена накоплением их в различных органах. Мукополисахариды играют важную роль в поддержании упругости и целостности соединительной ткани, упругости хряща. Мукополисахаридозы наследуются по аутосомно-рецессивному типу, за исключением синдрома Гунтера. Существует несколько типов заболевания, различающихся по биохимическому дефекту.
Симптомы
Мукополисахаридоз 1 типа – синдром Гурлера. Характеризуется дефицитом фермента альфа-идуронизады, что приводит к накоплению продуктов метаболизма в тканях и их эскреции с мочой. Во всех тканях расположены вакуолизированные клетки, содержащие лизосомы, переполненные мукополисахаридами. В клетках головного мозга накапливаются липиды.
Дети с синдромом Гурлера в момент рождения выглядят здоровыми и клинические проявления появляются на втором году жизни. При объективном обследовании выявляют гепатоспленомегалию, усиленный кифоз, постоянные выделения из носа, шумное дыхание. Появляются дисморфические изменения: выпуклый и нависающий лоб, плоский нос с запавшей переносицей, грубые и утолщенные губы, гипертелоризм. Волосы на голове жесткие и густые. Язык увеличен, зубы мелкие, ушные раковины деформированы. Голос хриплый. Туловище короткое, грудная клетка деформирована, выражен грудной и пояснично-грудной кифоз. Эпифизы длинных костей утолщены и расширены, окостенение нарушено. Имеются проявления порока сердца. Живот увеличен, имеют место гепатомегалия и пупочная грыжа. Пальцы кистей находятся в полусогнутом положении, ограничена подвижность суставов. У большинства больных наблюдается помутнение роговицы, катаракта, врожденная глаукома. Интеллект страдает, больные страдает, больные отстают в умственном развитии.
Мукополисахаридоз 2 типа – синдром Гунтера. Это единственный мукополисахаридоз, сцепленный с Х-хромосомой. Встречается у мальчиков. Течение его более доброкачественное. Развитие этого заболевания связано с недостаточностью фермента идуронатсульфатазы. Существует тип А и тип Б этого заболевания.
Тип А – классическая форма синдрома Гунтера. Для нее характерны грубые черты лица, низкорослость, тугоподвижность суставов, гепатоспленомегалия, грыжи, выраженная умственная отсталость. Заболевание прогрессирует медленнее. Часто у больных утрачен слух. Нередки кожные проявления, заключающиеся в появлении папул на коже спины. В патологический процесс часто вовлекается сердце. Может отмечаться умеренный кифоз. Продолжительность жизни – около 20 лет.
Тип Б – значительно более доброкачественный, продолжительность жизни у больных большая.
Мукополисахаридоз 3 типа – синдром Санфилиппо. Сопровождается эскрецией с мочой гепаринсульфата, который также накапливается в тканях. Заболевание проявляется самой глубокой прогрессирующей задержкой умственного развития. Клинические проявления ярко выражены у детей раннего возраста. Ребенок отстает в развитии, гиперактивен. До 10 лет идет быстрое прогрессирующее ухудшение состояния. Большинство детей умирает в середине второго десятилетия жизни. К характерным симптомам заболевания относятся умственная отсталость, тугоподвижность суставов, гепатоспленомегалия, грыжи, множественный дизостоз.
Мукополисахаридоз 4 типа – синдром Моркио. Обусловлен дефицитом ферментов гаактозо-6-сульфатазы и галактозамин-6-сульфатазы. На 2-ом году жизни дети начинают отставать в росте и у них появляется скелетные деформации (вальгусная деформация коленных суставов, выбухание нижних ребер, кифосколиоз). Степень укорочения туловища превышает укорочение конечностей. Интеллект относительно сохранен. Отмечается задержка физического развития, помутнение роговицы, выступающая нижняя часть лица, гипоплазия эмали зубов, короткая шея, килевидная грудная клетка, поясничный лордоз. У таких детей большой живот, гиперподвижность и подвывихи суставов, плоскостопие, отмечается снижение слуха. К 20 годам выявляется регургитация аорты. Смерть к этому возрасту обычно наступаетот сердечной и неврологической симптоматики (вследствие сдавления спинного мозга деформированными позвонками). Диагностика основана на выявлении в моче кератансульфата или кислых мукополисахаридов. Тип наследования аутосомно-рецессивный.
Ассоциированные симптомы: Агрессивность. Ограничение амплитуды движений.
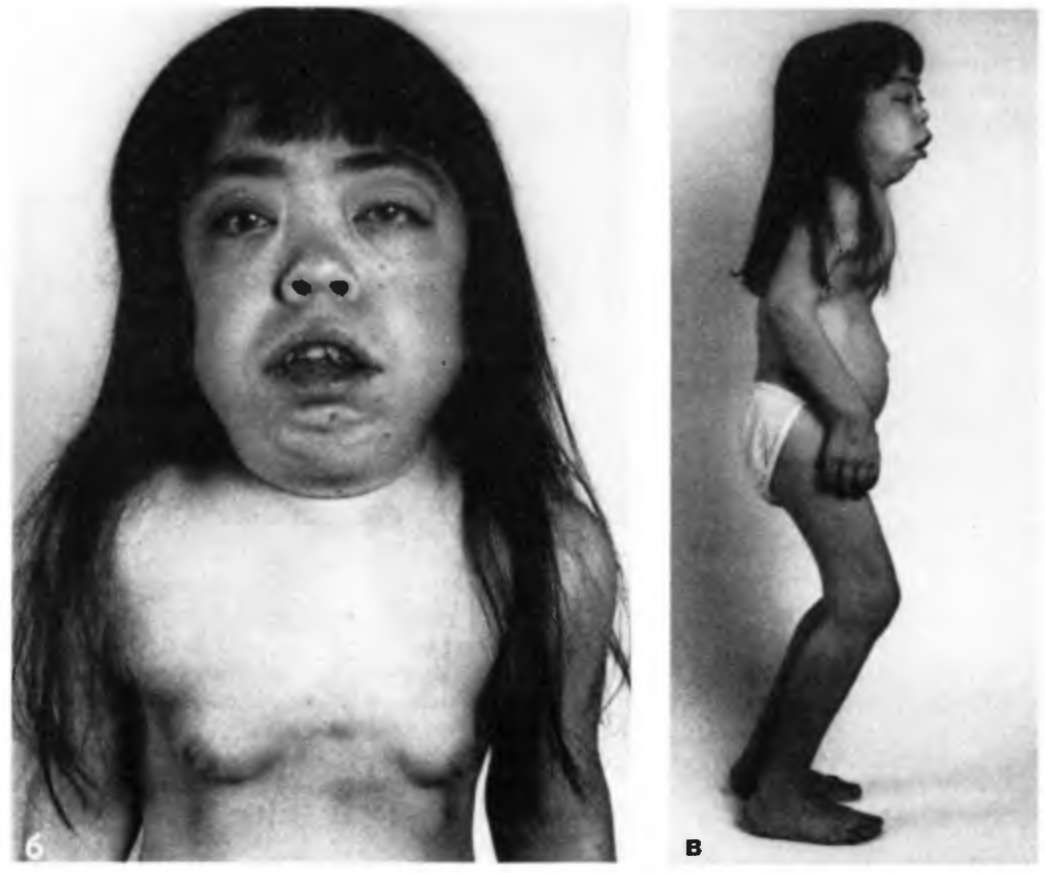
Внешний вид больной с мукополисахаридозом
Лечение
До сих пор не существует эффективного лечения синдрома Гурлера, в основном применяется симптоматическое и ортопедическое терапия. Больные обычно умирают в подростком возрасте.
Лечение заболевания заключается в назначении трансфузий крови, плазмы и лейкоцитарной массы. Показано применение глюкокортикоидов, тиреоидина, витамина А, витаминов группы В.
Прогноз
Прогноз при всех типах мукополисахаридозов неблагоприятный, болезнь неуклонно прогрессирует, и летальный исход может наступить в любом возрасте. Нередки случаи смерти больных от сопуствующей патологии сердца и присоединения инфекционного агента.
Профилактика
Так как в настоящее время нет эффективной терапии мукополисахаридозов, то необходимо выявлять заболевание в неонатальном периоде. Целесообразным представляется проведение трансабдоминального амниоцентеза у беременных женщин с высоким уровнем риска рождения детей с мукополисахаридозом. Культивирование клеток амниотической жидкости и определение в них активности ферментов, расщепляющих гликозамингликаны, позволяет проводить адекватную антенатальную диагностику мукополисахаридозов в ранние сроки беременности (9-12 недель). Не менее важным компонентом профилактики мукополисахаридозов представляется проведение селективного скрининга с целью обследования некоторых контингентов больных: людей с низким зрением, с задержкой умственного развития, нарушениями опорно-двигательного аппарата.
Основные медуслуги по стандартам лечения | ||
Клиники для лечения с лучшими ценами
|
Источник