
Операция на сердце с синдромом марфана

Синдром Марфана – аутосомно-доминантное наследственное заболевание, обусловленное мутацией гена, кодирующего синтез одного из базовых компонентов соединительной ткани – гликопротеида фибриллина. В частности, генетическое исследование при синдроме выявляет дефект гена FBN1, ответственного за синтез фибриллина-1 и входящего в состав коллагена. Следствием является замена в молекуле аминокислоты пролина на аргинин, что ведет к диспропорциональному превалированию в соединительной ткани коллагена III типа и характерным изменениям ее каркасно-эластических свойств в виде гиперрастяжимости.
Синдром имеет плейотропный характер с одинаковой распространенностью во всех этнических группах (1 случай на 5000) и классическим менделевским наследованием от больного родителя в большинстве случаев, однако у 25-30% пациентов возникает в виде первичной мутации.1 Все многообразие клинических проявлений непосредственно связано с системным вовлечением соединительной ткани. Главную опасность для жизни представляют собой изменения грудной аорты с неизбежным возникновением аневризм и расслоений, непосредственно определяющих прогноз течения заболевания.
Прогноз заболевания до появления хирургических методов лечения аневризм грудной аорты был неблагоприятным, и средняя продолжительность жизни пациентов с синдромом Марфана не превышала 30–40 лет. Летальные исходы в основном обусловлены острым расслоением и разрывом аневризм грудной аорты и/или развитием застойной сердечной недостаточности. В настоящее время в странах с развитым здравоохранением пациенты доживают до преклонного возраста.5
Клинические проявления синдрома Марфана в манифестированных случаях довольно характерны и включают в себя:
- Скелетные аномалии: высокий рост, астеническое телосложение, долихостеномелия (непропорционально длинные руки и ноги), арахнодактилия, воронкообразная или килевидная деформация грудной клетки, кифосколиоз, плоскостопие, долихоцефалия, узкий лицевой скелет («птичье» лицо), «готическое» нёбо. Характерна гиперподвижность суставов. Мышцы часто отстают в росте от скелета, развиты слабо.
- Поражение сердечно–сосудистой системы: патогномонично расширение восходящей аорты, острое расслоение грудной аорты и пролапс митрального клапана.
- Патология зрения: чаще наблюдается миопия вследствие увеличения длины глазного яблока, но возможна гиперметропия. Слабость связочного аппарата хрусталика приводит к его подвывиху или полному вывиху (дислокация хрусталика), что может сопровождаться дрожанием радужной оболочки (иридодонез). Развиваются вторичная глаукома, отслойка сетчатки, катаракта. Характерны голубые склеры.
- Иные полиморфные клинические проявления в виде эмфиземы и спонтанного пневмоторакса за счет разрыва легочных «булл» (чаще у взрослых), гастроптоз, дискинезия желудочно–кишечного тракта, нефроптоз, бедренные, паховые и диафрагмальные грыжи, гипоплазия мышц и подкожной клетчатки, мышечная гипотония и др.
- Психоневрологические нарушения в виде повышенной нервной возбудимости, астено–невротического синдрома, эмоционально-волевых нарушений.
Диагностика
Основывается на Гентских критериях 2010 г. и подтверждается углубленным генетическим анализом с обнаружением дефекта гена FBN1.3
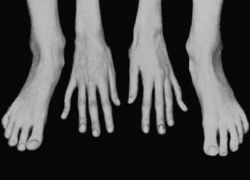
Рис. 1 Характерные проявления синдрома Марфана: длинные и тонкие пальцы (арахнодактилия)
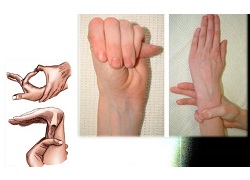
Рис. 2 Диагностические тесты (тест запястья, тест большого пальца)
Клинический случай
Пациентка Б. 1998 года рождения (21 год) поступила в отделение кардиохирургии НМИЦ хирургии им. А.В. Вишневского 31 мая 2019 года. Активно жалоб при поступлении не предъявляет, при детальном расспросе – жалобы на боли в грудной клетке неясной локализации, четко не связанные с физической активностью, одышку, слабость. Из семейного анамнеза известно, что мать пациентки умерла в возрасте 30 лет от острого расслоения аорты. Пациентка с 6 лет была на учете у педиатра с подозрением на синдром Марфана в связи с ранним развитием симптомов данного заболевания: астеническое телосложение, сколиоз грудного отдела позвоночника. С 14 лет находится на учете у кардиолога по поводу диагностированного расширения восходящего отдела аорты. В 15–летнем возрасте генетически подтвержден синдром Марфана, выявлен дефект гена FBN1. С 19–летнего возраста отмечается снижение остроты зрения, миопия слева (–0,75), справа (–1,5).
Ежегодно проводился контроль посредством трансторакальной эхокардиографии и мультиспиральной компьютерной томографии с целью оценки размеров и состояния корня и грудного отдела аорты. До 20–летнего возраста диаметр корня и грудного отдела стабильный, значительного роста этих показателей не отмечается. С 20–летнего возраста наблюдается отрицательная динамика с прогрессивным увеличением размеров восходящего отдела аорты до 44 мм. В 2019 году отмечено резкое увеличение данного сегмента аорты с 45 мм до 50 мм. Дополнительно при МСКТ исследовании в июне 2019 года выявлена локальная диссекция некоронарного синуса аорты.
Также из анамнеза известно, что пациентка в 14–летнем возрасте перенесла субтотальную резекцию щитовидной железы по поводу диффузно–токсического зоба и находится на заместительной терапии L–тироксином 100 мкг.
При осмотре: телосложение астеническое, рост 178 см, вес 65 кг, индекс массы тела 21, имеются лицевые дизморфии (долихоцефалия, гипоплазия скуловых костей, энофтальм), подкожная жировая клетчатка развита слабо. Выявляется левосторонний грудопоясничный сколиоз, умеренная воронкообразная деформация грудной клетки. Отношение длины верхней половины туловища к нижней 0,80, размаха рук к росту 1,17. При аускультации выслушивается диастолический шум во II точке.
Эхокардиографическое исследование выявило умеренную недостаточность аортального клапана 2–2,5 степени, ширина струи регургитации при этом 3,5 мм. Структура створок аортального клапана сохранена, максимальный градиент 4 мм рт.ст. Корень аорты расширен до 38 мм. Митральный и трикуспидальный клапан в пределах нормы. Размеры полостей сердца и насосная функция в пределах нормы, имеется умеренная гипертрофия левого желудочка.
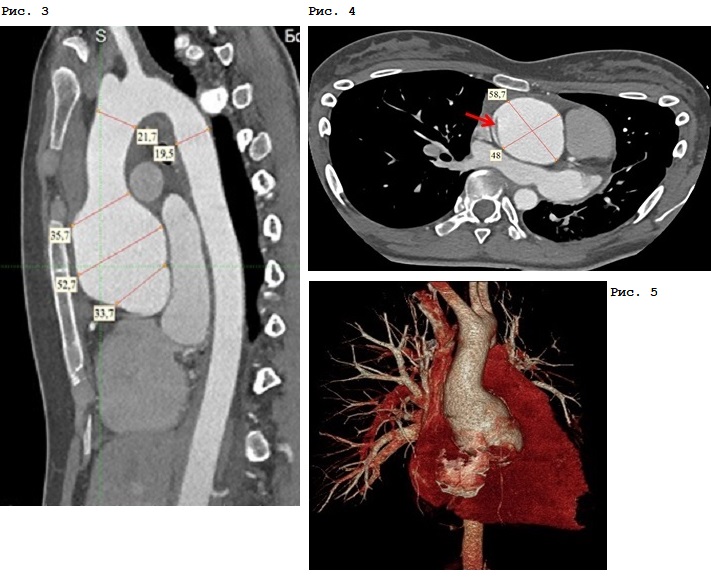
По данным МСКТ грудного отдела аорты с контрастированием наблюдается расширение фиброзного кольца аортального клапана до 32 мм, и тотчас, начиная с уровня синусов Вальсальвы, отмечается расширение аорты до 53 мм, в области синотубулярного соединения до 58х48 мм (рис. 3, красными линиями указаны линейные размеры корня и восходящего отдела аорты). При контрольном исследовании от 03.06.19 по правой и левой полуокружностям восходящего отдела аорты на 53 мм и 32 мм дистальнее фиброзного кольца определяются участки расслоения аорты (рис. 4, красными линиями указаны размеры восходящего отдела аорты; красной стрелкой – зона диссекции). Дуга аорты размерами до 22 мм, не изменена, нисходящий отдел грудной аорты – до 20 мм, также без патологических изменений (рис. 5, 3D–реконструкция грудного отдела аорты).
Диагноз синдрома Марфана был очевиден на основании Гентских критериев, включая семейный анамнез, наличие признаков системного вовлечения соединительной ткани (воронкообразная деформация грудной клетки, кифосколиоз, лицевые дизморфии), наличие аневризмы грудной аорты.
Согласно Рекомендациям по диагностике и лечению заболеваний аорты Европейского общества кардиологов (ESC) от 2014 г., показания к хирургическому вмешательству должны основываться как на размерах аорты, так и особенностях течения заболевания, и риск операции не должен превышать риска его естественного течения. Показания к операции при синдроме Марфана возникают при размерах аорты ≥50 мм. В случае размеров аорты 45–50 мм рассматривается наличие дополнительных факторов риска, таких как семейный анамнез расслоения аорты, быстрый рост диаметра аорты > 3 мм в год и значимая регургитация на аортальном клапане.
Показания к операции в данном клиническом наблюдении не вызывали сомнений, поскольку риск разрыва и расслоения аорты был крайне высокий. Выбор же самой стратегии вмешательства носил дискутабельный характер. Возможно было применить операцию протезирования корня аорты с полным замещением искусственным клапан–содержащим кондуитом или реконструктивную клапан–сохраняющую операцию – с возможностью реимплантации нативного аортального клапана или ремоделирования корня аорты с сохранением аортального клапана.
Операция Бенталла–Де Боно (Bentall–De Bono), или полное замещение корня и восходящего отдела аорты клапан–содержащим кондуитом (рис. 6), – хорошо зарекомендовавшее себя, надежное вмешательство в аортальной хирургии. Имеет ряд очевидных преимуществ, таких как гарантированное длительное функционирование механического протеза и меньшая сложность техники операции. К негативным факторам можно отнести необходимость пожизненного приема антикоагулянтов с постоянным контролем антикоагулянтной терапии. Любая отмена терапии может привести в дисфункции протеза с тяжелыми последствиями. Соответственно, отсутствует возможность беременности и родов.
У молодых женщин с необходимостью сохранения детородной функции возможно применение стратегии клапан–сохраняющих операций в двух вариантах: ремоделирование корня аорты, или операция Якуба (Yacoob) (рис. 7) и реимплантация аортального клапана, или операция Дэвида (David) (рис. 8) в ее модификациях.
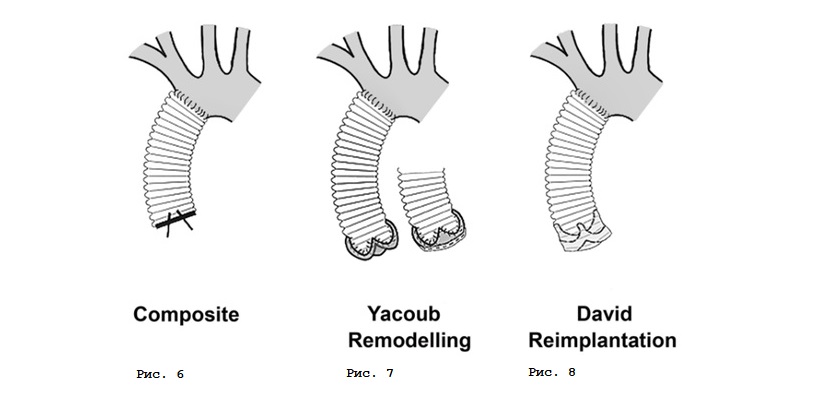
Суть операция Якуба заключается в том, что аортальные синусы заменяются специально смоделированным в виде короны сосудистым протезом таким образом, что сохраняется функциональная связь корня аорты и створок аортального клапана. Второй вариант, более безопасный и воспроизводимый, – операция Дэвида, существующая в нескольких модификациях (I, II, III, IV, V). На сегодняшний день чаще всего используется модификация David V. Суть – в имплантации всех структур аортального клапана внутрь сосудистого протеза с сохранением анатомо–функционального взаимоотношения комиссур и створок аортального клапана. Методику операции отличает надежность гемостаза и функции аортального клапана.
Основным преимуществом обеих клапан-сохраняющих процедур является сохранность нативного клапана, что обеспечивает его хорошую функцию и позволяет отказаться от приема антикоагулянтов.
В нашем случае теоретически имелся риск дисфункции аортального клапана вследствие возможной его некомпетенции в отдаленном периоде по причине системной неполноценности соединительной ткани, в том числе структур самого клапана.
Дискуссия о возможности клапан–сохраняющих операций при синдроме Марфана подошла к завершению только в последние годы после публикации нескольких крупных исследований. В частности, A. Martens с соавт. опубликовали данные 20–летнего наблюдения более сотни пациентов с синдромом Марфана, которым была выполнена процедура David. Авторы пришли к выводу, что клапан–сохраняющие процедуры на корне аорты должны быть рассмотрены именно в качестве предпочтительного метода лечения аневризмы корня аорты у пациентов с синдромом Марфана при сохранных створках клапана. При этом, по–видимому, соединительно–тканная дисфункция не затрагивает структуры аортального клапана и таким образом не имеет негативного влияния на отдаленные результаты вмешательства. Долговечность клапан–сохраняющих процедур при синдроме Марфана обусловлена исключительно правильным отбором пациентов и качеством самой операции. Этим авторы объясняют разнородность полученных ранее результатов в других исследованиях и настаивают на необходимости строгой индивидуальной оценки клапана и достаточного опыта данного вмешательства.8
Еще одно исследование, сравнивающее результаты операций David и Bentall–De Bono у пациентов с синдромом Марфана, опубликовано J. Price с соавт. В частности, сравниваются клапан–сохраняющие процедуры у 98 больных с операцией Bentall–De Bono – у 67. В отдаленном периоде в группе операций Bentall чаще возникали расслоения аорты (25.4 vs 4.1%), умеренная или тяжелая аортальная недостаточность (49.3% vs 14.4%), а также чаще регистрировались экстренные оперативные вмешательства (24.6% vs 3.3%). Госпитальной летальности не было, в отдаленном периоде в сроки до 17 зафиксировано 9 летальных исходов. Соответственно 10–летняя выживаемость в группе Bentall была 90.5%, а в группе клапан–сохраняющих вмешательств – 96.3%. Кроме того, клапан–сохраняющие вмешательства были ассоциированы с меньшим риском тромбоэмболических и геморрагических осложнений, но отдаленная выживаемость и свобода от повторных операций, а также риск вторичного эндокардита в обеих группах были сопоставимы.9
Учитывая вышеперечисленное, нами была выбрана методика клапан–сохраняющей процедуры (табл. 1).
Таблица 1. Преимущества и недостатки стратегий операций вмешательств.
Клапан-сохраняющие процедуры | Протезирование корня аорты | |
Преимущества |
|
|
Недостатки |
|
|
17 июня 2019 г. у данной пациентки нами была выполнена операция Дэвида. Доступ: срединная стернотомия. Канюляция аорты в средней трети дуги аорты канюлей диаметром 21 Fr с использованием методики Сельдингера. Раздельная канюляция ВПВ канюлей 24 Fr. Нижняя полая вена канюлирована через ушко правого предсердия канюлей 32 Fr.
Искусственное кровообращение в условиях умеренной гипотермии до 29 градусов С в носоглотке. Дренирование левого желудочка через правую верхнюю легочную вену. Восходящая аорта пережата на 1 см проксимальнее устья брахиоцефального ствола под контролем церебральной оксиметрии. Кардиоплегия раствором «Кустодиол» антеградно селективно в устья коронарных артерий (1000 мл в левую коронарную артерию, 1000 мл в правую коронарную артерию). Подготовка аортального клапана к реимплантации: выделен корень аорты, вскрыт просвет аорты, выполнена ревизия аортального клапана. На комиссуры наложены швы–держалки, иссечена стенка аорты в зоне коронарных синусов, выделены устья коронарных артерий на «кнопках». Использован специально сформированный в виде синусов Вальсальвы синтетический протез BBraun Sinus 28 мм. Выполнена реимплантация аортального клапана в сосудистый протез: наложены швы на уровне фиброзного кольца изнутри наружу синтетической атравматической полиэстерной нитью 2–0 с прокладками с прошиванием проксимального края сосудистого протеза, произведена фиксация сосудистого протеза к фиброзному кольцу. Повторный пассаж кардиоплегии по 500 мл в устья коронарных артерий. Далее – моделирование высоты комиссур по отношению к сосудистому протезу для получения оптимальной коаптации аортальных створок, формирование гемостатических швов клапана, затем реимплантация устьев коронарных артерий на «кнопках» в модификации Kouchukos. Заключительный пассаж кардиоплегии. Между аортой и отдельным протезом 28 мм сформирован дистальный анастомоз по методике «погружения» внутрь аорты на 2 см проксимальнее брахиоцефального ствола. Проксимальный и дистальный протез сшиты между собой. Для дополнительной герметизации швом был использован хирургический клей BioGlue. Стандартное окончание ИК и операции (рис. 11).
Рис. 11 Схематичное изображение хода операции
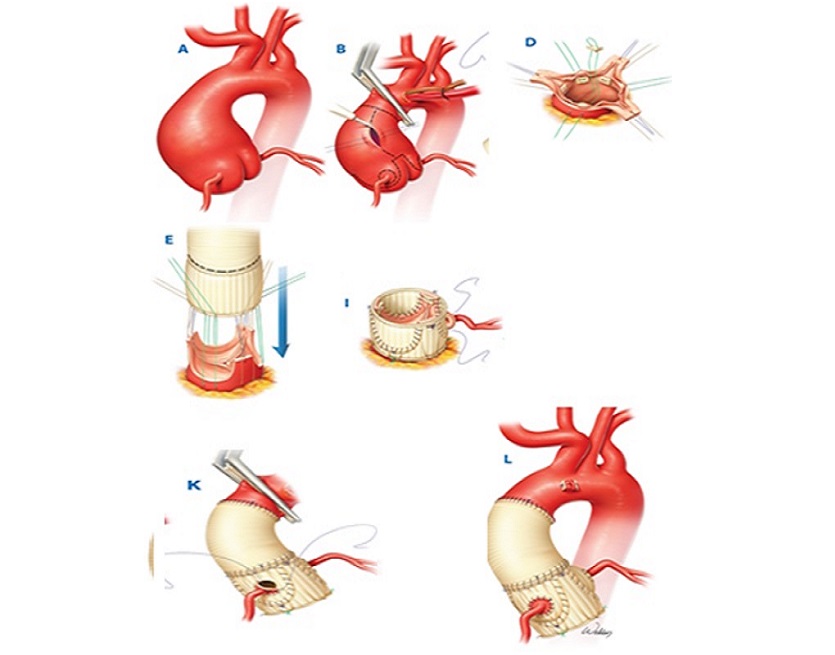
Пациентка переведена в отделение реанимации на искусственной вентиляции легких, время искусственной вентиляции легких составило 15 часов, время пребывания в ОРИТ – 76 часов. Количество отделяемого по дренажам за первые сутки – 450 мл, за вторые сутки – 100 мл. На 4–е послеоперационные сутки пациентка в удовлетворительном состоянии переведена в профильное отделение.
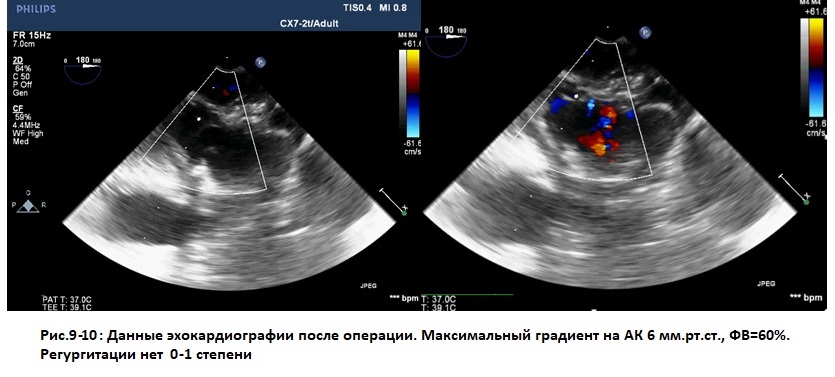
Послеоперационный период протекал без особенностей, послеоперационные раны заживали первичным натяжением, грудина стабильна. На 14-е послеоперационные сутки пациентка выписана под наблюдение кардиолога по месту жительства. По данным трансторакальной ЭХОКГ в послеоперационном периоде функция клапана удовлетворительная, максимальный градиент на аортальном клапане 6 мм рт.ст., регургитация до 1 ст. (рис. 9). По данным МСКТ с контрастированием, протез корня аорты на протяжении 48 мм, диаметр до 30 мм, восходящий отдел аорты до 21 мм, проксимальный отдел аорты до 21 мм (рис. 10).
Выводы
Современные технологии операций на корне аорты с сохранением нативного аортального клапана позволяют обеспечить благоприятные непосредственные функциональные результаты вмешательства и в перспективе могут улучшить прогноз и качество жизни у пациентов с синдромом Марфана и аневризмой восходящей аорты.
Литература
1. Медицинская генетика: учебник // В. Н. Запорожан, Ю. И., Бажора, А. В. Шевеленкова, М. М. Чеснокова. — Одесса: ОНМедУ, 2012. — 278 с. — (Серия «Библиотека студента–медика»). ISBN 978–966–443–004–0
2. Международные рекомендации диагностики синдрома Марфана — Гентские критерии (Ghent criteria, De Paepe A. et al., 1996, Loeys B. et al., 2010)
3. Всероссийское научное общество кардиологов. «Наследственные нарушения соединительной ткани», российские рекомендации, секция «Дисплазии соединительной ткани сердца», Москва, 2012
4. «Диагностика и лечение наследственных и многофакторных нарушений соединительной ткани». Национальные клинические рекомендации, Республика Беларусь, Минск, 2014 г.
5. Клиническая генетика: учебник // Г.Р.Мутовин – ГЭОТАР–Медиа, 2010 г.
6. David, T. E. (2016). Aortic Valve Sparing in Different Aortic Valve and Aortic Root Conditions. Journal of the American College of Cardiology, 68(6), 654–664.doi:10.1016/j.jacc.2016.04.062
7. 2014 ESC Guidelines on the diagnosis and treatment of aortic diseases; The Task Force for the Diagnosis and Treatment of Aortic Diseases of the European Society of Cardiology (ESC)
8. Valve–sparing aortic root replacement in patients with Marfan syndrome-the Homburg experience// Ulrich Schneider, Tristan Ehrlich, Irem Karliova, Christian Giebels, Hans–Joachim Schäfers. Department of Thoracic and Cardiovascular Surgery, Saarland University Medical Center, Homburg/Saar, Germany
9. Long–term outcomes of aortic root operations for Marfan syndrome: A comparison of Bentall versus aortic valve–sparing procedures.// Joel Price, MD, et al – J Thorac Cardiovasc Surg. 2016 Feb;151(2):330–6. doi: 10.1016/j.jtcvs.2015.10.068. Epub 2015 Oct 27
10. Valve–Sparing Aortic Root Replacement: Early and Midterm Outcomes in 83 Patients. J.Coselli et al. The Annals of Thoracic Surgery, April 2014 Volume 97, Issue 4, Pages 1267–1274
Источник
Интервью: Ксения Акиньшина
О СУЩЕСТВОВАНИИ ГЕНЕТИЧЕСКИХ ЗАБОЛЕВАНИЙ СЛЫШАЛИ ВСЕ, но мало кто представляет, как их много. Не все выявляются при рождении: например о синдроме Марфана становится известно гораздо позже. Это генетическое заболевание, при котором нарушается синтез белка фибриллина — он отвечает за эластичность и сократимость соединительной ткани. Поражаются многие системы и ткани организма: сосуды и сердце, кости и суставы, глаза и лёгкие. Люди с этим заболеванием обычно высокие, с длинными руками, ногами, кистями и стопами; их вытянутые пальцы в медицинских учебниках принято называть «паучьими». Лечение и прогноз напрямую зависят от степени тяжести: например, если поражена аорта — самый большой сосуд в организме человека — состояние становится угрожающим жизни. Мы поговорили со Светланой Х. о том, как она живёт с синдромом Марфана.
Мне тридцать лет, а о диагнозе стало известно, когда мне было шесть. Я быстро росла, и на очередном плановом осмотре врач услышал шумы в сердце; после этого был приём у генетика и предположительный диагноз — синдром Марфана; в статусе «предположительного» он оставался многие годы. Болезнь тогда была плохо изучена, и единственными рекомендациями были наблюдение кардиолога и запрет на физические нагрузки. Потом, правда, стало понятно, что необходима лечебная физкультура, и я пошла на плавание. Получалось хорошо: я занималась в школе олимпийского резерва и в девять лет меня даже пригласили в профессиональную команду. Кардиолог и родители, правда, были против увеличения нагрузок — расстроившись, я бросила плавание вообще.
По мере взросления проблемы с сердцем ухудшались: я росла, а вместе со мной растягивалась аорта — самый большой сосуд организма. Уже в детском возрасте все параметры аорты превышали нормальные значения даже для взрослого человека. Проблемы с клапанами сердца тоже шли по нарастающей. В десять лет мне довелось побывать на консилиуме врачей — в то время это было редкостью, да ещё в бесплатной медицине, то есть случай был тяжёлым. Решался вопрос кардиохирургии, но прямых показаний к операции не было — просто «вялотекущее ухудшение», да и «неизвестно, как поведут себя ткани после операции, может быть, будут расползаться». Генетически подтвердить или опровергнуть диагноз тогда не предлагали — то ли врачи были не в курсе такой возможности, то ли её тогда просто не существовало.
Течение болезни бывает очень тяжёлым, и к лечению нужно подходить ответственно, поскольку на кону не только качество жизни, но и она сама: по статистике 90–95 % пациентов не дотягивают до 40–50 лет
В общем, единственными обследованиями были ЭКГ, ЭХО, холтеровское мониторирование (круглосуточная ЭКГ в условиях обычной повседневной активности, когда датчики ЭКГ приклеивают к телу. — Прим. ред.), посещения кардиолога, поддерживающая терапия и совет «не болеть». Следовать ему у меня не очень-то получалось. В тринадцать лет, пролежав дома две недели с температурой под сорок, я всё же попала в инфекционную больницу — и в приёмном отделении врач, увидев моё горло, побледнела и объявила моей маме, что у меня дифтерия. Мама расплакалась, а меня перевели в отделение и поместили в палату с выздоравливающими от гриппа — «чудесное» решение. Хорошо, что диагноз не подтвердился и дифтерии у меня всё же не было. Тем не менее любая болезнь, тем более поражающая дыхательные пути, негативно отражается на сердце, что в моём случае просто опасно.
Всю жизнь я прожила с диагнозом «дисплазия соединительной ткани», а синдром Марфана был лишь фенотипом — это значит, что у меня были проявления синдрома, но он не был подтверждён генетически. По мнению некоторых врачей, особенно тех, кто видел меня впервые, он отсутствовал вовсе — ведь полного набора классических симптомов не было. У меня всё более или менее нормально с костями и состоянием позвоночника, на месте хрусталик глаза; болезнь можно заподозрить только из-за патологий сердечно-сосудистой системы, высокого роста, арахнодактилии (те самые «паучьи пальцы») и повышенной эластичности. Я и сейчас, рассказывая врачам об анамнезе, упоминаю дисплазию чаще, чем синдром Марфана — иначе они просто вежливо кивают и пропускают мимо ушей.
Поскольку меня не беспокоила боль, я жила как обычный подросток. За моё сердце переживала мама, но неполная осведомлённость помогла пройти этот путь гораздо легче. Если бы тогда она была в курсе всех сюрпризов, которые может преподнести болезнь, не знаю, как справлялась бы с этим. Я даже рада, что и сейчас она знает не больше прежнего — теперь уже я думаю о её сердце. Реально же подтвердить диагноз удалось только на тридцатом году жизни: я самостоятельно сдала анализ на мутации генов в рамках так называемой панели заболеваний соединительной ткани. С его помощью выявили мутацию, характерную для синдрома Марфана, однако не в «горячих точках» — возможно, поэтому я и не типичный представитель болезни.
Синдром Марфана — это генетическое заболевание, но оно не обязано проявляться у кого-то из родни. У моих родителей, например, нет никаких проявлений, ни внешне, ни «внутренне». В основе заболевания лежат мутации в гене, отвечающем за синтез фибриллина — важнейшего структурного белка межклеточного матрикса, придающего эластичность и сократимость соединительной ткани. При нём сильнее всего страдают органы с наибольшей «концентрацией» соединительной ткани: сердце, глаза, спина, связки. Синдром не лечится, и любая терапия направлена на конкретные пострадавшие органы — например, я должна пить курсами таблетки, помогающие сердечно-сосудистой системе. Течение болезни бывает очень тяжёлым, и к лечению нужно подходить ответственно, поскольку на кону не только качество жизни, но и она сама: по статистике 90–95 % пациентов не дотягивают до 40–50 лет.
Ограничения в первую очередь касаются физических нагрузок. Нельзя заниматься профессиональным спортом, хотя по иронии судьбы данных у меня как раз предостаточно — например, высокий рост и колоссальная гибкость (до сих пор могу закинуть ногу за голову или сесть в позу лотоса с разбега). Людям с синдромом Марфана показан разумный спорт без резких движений, например плавание. В путешествиях моя аптечка не больше, чем у обычного человека, никакой специфики — но тут дело скорее не в факте синдрома, а в степени его проявления.
Из-за «кукольного театра», как назвали ширму между мной и оперирующими врачами, я слышала, как акушер постоянно повторяла ассистентам: «Не рвите ткани, не рвите ткани!»
Со всей серьёзностью проблемы я столкнулась, когда мы с мужем захотели ребёнка. Генетик приговорил к суррогатному материнству или возможной беременности после кардиохирургии, к которой не было прямых показаний. Мне объясняли, что функции сердца достаточно для моего собственного жизнеобеспечения, но при беременности нагрузка удваивается. Я же настаивала, что при относительно лёгкой степени поражения сердца и сосудов нормальное течение беременности возможно. Я побывала во всех кардиоцентрах Москвы, пролила море слёз и всё же получила разрешение, при условии постоянного наблюдения кардиолога. Так, через два месяца я пришла вставать на учёт в кардиологический перинатальный центр, который согласился меня вести.
Конечно, решиться на беременность было очень страшно, особенно после неоднозначного прогноза врачей — моя аорта находится пока на докритическом уровне. Надеюсь, там она и останется — до критического лишь несколько миллиметров. Надежду вселила врач-гинеколог, посвятившая полжизни изучению дисплазии соединительной ткани. После разговора с ней я поняла, что если я не попробую выносить ребёнка вопреки своему желанию, то буду жалеть всю жизнь. Где-то глубоко внутри я взяла на себя этот риск и не жалею.
Беременность прошла отлично, а дочь родилась через плановое кесарево сечение немного раньше срока. Врачи безумно боялись, что аорта «рванёт» от максимальных нагрузок и я умру прямо в роддоме, где была самой «тяжёлой» роженицей. Дочь — моя победа, и мы назвали её Викторией. Из-за «кукольного театра», как назвали ширму между мной и оперирующими врачами, я слышала, как акушер постоянно повторяла ассистентам: «Не рвите ткани, не рвите ткани!» — но в тот момент я была готова ко всему, лишь бы с ребёнком всё было в порядке. Операция длилась в два раза дольше обычного, врач была мокрой, будто на неё вылили ведро воды. Я пару раз почти теряла сознание, в чувства приводил анестезиолог, а потом, уже в реанимации, я узнала, что потеряла почти литр крови. Состояние моей сердечно-сосудистой системы осталось без изменений, то есть таким же, как до беременности.
Оглядываясь на свою юность, я понимаю, что мне в какой-то степени повезло, если можно так выразиться. Да, меня не обошли высокий рост, пластинка на зубах, одно плечо выше другого — конечно, я выделялась из толпы, были насмешки среди сверстников и слёзы ночами в ванной комнате. Но такое было у многих, такова подростковая жизнь. После общения с мамами детей с более тяжёлым синдромом Марфана, я поняла, что моя жизнь могла бы быть гораздо труднее.
Свой оптимизм я взрастила сама, по крупинкам — при этом я заядлый параноик, что вообще присуще людям с синдромом Марфана. В интернете есть множество статей и информации, в которой очень легко запутаться и накрутить себя ещё сильнее. Специалистов же, которые разбираются в проблеме, единицы. Есть группы и форумы, где люди описывают свои симптомы, делятся опытом и даже «ставят» себе и другим диагнозы; многие хорошо изучили проблему и дадут в этой области фору некоторым докторам. Но большинство пишут о неизбежности, о моральной и физической боли, о доживании — поэтому я в этих группах не сижу, не хочу загонять себя в переживания ещё глубже. Конечно, я понимаю свои перспективы, но всегда ищу положительное в окружающем мире, стараюсь не зацикливаться на проблемах — иначе крайне сложно выбраться из разрастающейся паники. Конечно, не нужно закрывать глаза на свой диагноз, будто его нет — он есть, и очень опасен, но это не клеймо и не приговор.
О моей особенности знают лишь самые близкие, и многие люди из окружения задают вопросы о втором ребёнке. Но я не могу решиться на этот шаг, просто не имею права. Ещё до рождения дочери я взяла на себя колоссальную ответственность за неё и перед ней. Возможно, мне придётся столкнуться с кардиохирургией, и мне очень страшно. Мой организм с годами даёт о себе знать всё больше, количество визитов к врачам ежегодно растёт — но это не повод сидеть и считать оставшиеся дни. Мне бывает очень сложно. Мысли о судьбе дочери, которой уже два года, и о продолжительности собственной жизни порой неделями не дают спать, но я делаю всё возможное, чтобы минимизировать проявления синдрома — ведь если я сдамся, лучше не станет. Нужно жить свою жизнь, а не проживать её.
Фотографии: spacedrone808 — stock.adobe.com (1, 2, 3)
Источник