
Синдром прадера вилли спв в

Что такое синдром Прадера — Вилли?
Синдром Прадера — Вилли (СПВ) — это генетическое заболевание, которое встречается примерно у одного из каждых 15 000 новорожденных.
Синдром Прадера — Вилли поражает мужчин и женщин с одинаковой частотой и затрагивает все расы и этнические группы. СПВ признана наиболее распространенной генетической причиной опасного для жизни детского ожирения.
Данный синдром был впервые описан швейцарскими врачами Андреа Прадером, Алексисом Лабхартом и Генрихом Вилли в 1956 году на основании клинических характеристик девяти обследованных детей.
Общие характеристики, определенные в первоначальном отчете, включали маленькие руки и ноги, аномальный рост и состав тела (маленький рост, очень низкая мышечная масса тела и раннее детское ожирение), мышечная гипотония (низкий мышечный тонус (слабость мышц)) при рождении, ненасытный голод, сильное ожирение и умственная отсталость.
Это видео даст краткое понимание проявлений болезни:
Синдром Прадера — Вилли является результатом аномалии хромосомы 15, и постановка точного диагноза сегодня основывается на генетических анализах.
Каковы симптомы синдрома Прадера — Вилли?
Симптомы синдрома Прадера — Вилли, вероятно, связаны с дисфункцией части мозга, называемой гипоталамус. Гипоталамус — это небольшой эндокринный орган у основания мозга, который играет важную роль во многих функциях организма, включая регулирование голода и сытости, температуры тела, боли, баланса сна и бодрствования, водного баланса, эмоций и фертильности.
Хотя считается, что дисфункция гипоталамуса приводит к появлению симптомов синдрома Прадера — Вилли, еще не ясно, как генетическая аномалия вызывает дисфункцию гипоталамуса.
У людей с данным синдромом симптомы со временем меняются. В целом, есть два основных этапа симптомов, связанных с СПВ:
Ранний период жизни
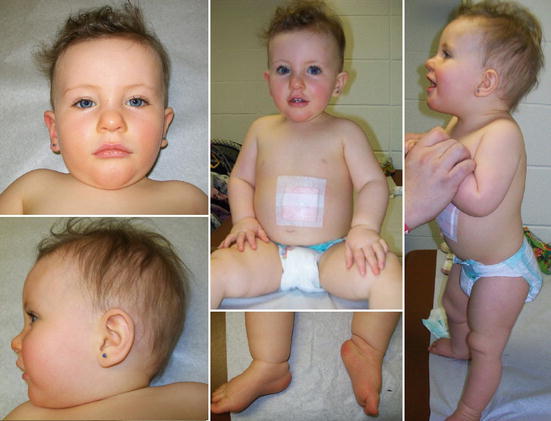
Младенцы с СПВ являются гипотоническими или «гибкими», с очень низким мышечным тонусом. Для них типичен слабый крик и плохой сосательный рефлекс. Детей с синдромом Прадера — Вилли обычно не могут кормить грудью и они часто нуждаются в искусственном вскармливании.
У детей могут быть проблемы с ростом и развитием, если трудности с кормлением не подвергаются тщательному мониторингу и лечению. По мере взросления этих детей сила и мышечный тонус в целом улучшаются. Моторные функции развиты, но, как правило, выполняются с задержкой.
Малыши, как правило, вступают в период, когда они могут легко начать набирать вес до того, как проявят повышенный интерес к еде.
Детство и взросление
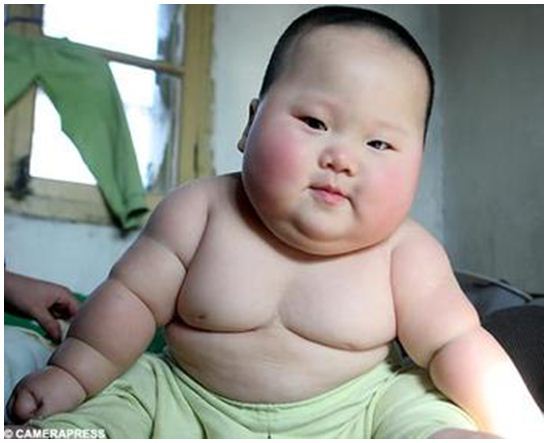
Нерегулируемый аппетит и легкое увеличение веса (см. фото) характеризуют более поздние стадии синдрома Прадера — Вилли.
Эти признаки чаще всего начинаются в возрасте от 3 до 8 лет, но различаются по началу и интенсивности.
Люди с синдромом Прадера — Виллии испытывают недостаток в нормальных подсказках голода и сытости.
Как правило, они не могут контролировать потребление пищи и переедают, если их не контролировать.
Поведение в голоде очень распространено. Кроме того, скорость метаболизма у людей с данным синдромом ниже, чем обычно. Без лечения эта комбинация проблем приводит к патологическому ожирению и его многочисленным осложнениям.
Помимо ожирения, у ребенка с синдромом Прадера — Вилли может быть множество других симптомов. Дети с IQ от низкого нормального до умеренной умственной отсталости обычно имеют когнитивные проблемы. Те, у кого нормальный IQ, обычно имеют проблемы с обучаемостью.
Другие проблемы и признаки могут включать:
- дефицит гормона роста/низкий рост;
- маленькие руки и ноги;
- сколиоз;
- нарушения сна с чрезмерной дневной сонливостью;
- высокий болевой порог;
- апраксия/диспраксия речи и бесплодие.
Поведенческие трудности могут включать в себя обсессивно-компульсивные симптомы, проблемы с кожей и трудности с контролем эмоций. Взрослые с синдромом Прадера— Вилли подвергаются повышенному риску психических заболеваний. СПВ является расстройством широкого спектра, и симптомы варьируют по степени тяжести и частоты встречаемости среди людей.
Хромосомы и гены: основы
Чтобы понять генетику СПВ, необходимо иметь общее представление о хромосомах и генах.
Хромосомы представляют собой крошечные структуры, которые присутствуют почти в каждой клетке нашего тела. Это пакеты генов, которые мы наследуем от наших родителей. Гены содержат все подробные инструкции, необходимые нашему телу для роста, развития и функционирования — нашу ДНК.
Определенные гены направляют наши клетки на производство белков, ферментов и других необходимых веществ. Каждый из наших многочисленных генов находится на определенной хромосоме. Большинство клеток нашего тела содержат 46 хромосом — 23 унаследованы от нашей матери и 23 от нашего отца. (Яйца и сперматозоиды обычно содержат всего 23 хромосомы, потому что это те клетки объединяются в зачатии и обеспечивают ребенку нужное количество хромосом.)
22 пары хромосом помечены числом, основанным на их размере (хромосома 1 — самая большая пара, а хромосома 22 — почти наименьшая), и 2 хромосомы в каждой пронумерованной паре содержат одинаковые гены (один набор у матери и одну от отца). Изменения, которые вызывают синдром Прадера — Вилли, происходят в паре, известной как хромосома 15. 23-я пара хромосом обозначена как пара половых хромосом. Эта пара определяет пол ребенка: XX для девочки, XY для мальчика.
Изменения или ошибки в генах и хромосомах распространены при образовании яйцеклеток и сперматозоидов. Некоторые из этих генетических изменений не будут иметь эффекта, когда ребенок зачат; некоторые вызовут выкидыш; а некоторые спровоцируют, например, синдром Прадера — Вилли, вызвав значительные различия в том, как ребенок развивается и функционирует.
В то время как многие генетические нарушения вызваны изменением одного гена и могут передаваться от родителя к ребенку, СПВ куда более сложен.
Что вызывает синдром Прадера — Вилли (причины)?
Синдром Прадера — Вилли возникает в результате отсутствия активного генетического материала в определенной области хромосомы 15 (15q11-q13). Обычно люди наследуют одну копию хромосомы 15 от своей матери и одну от своего отца. Гены вызывающие СПВ активны только в хромосоме, полученной от отца.
При синдроме Прадера — Вилли генетический дефект, вызывающий неактивность хромосомы 15 от отца (отцовская хромосома 15), может возникнуть по трем следующим причинам:
- СПВ по исключению: чаще всего часть хромосомы 15, которая была унаследована от отца, отсутствует или удалена в этой критической области. Эта небольшая делеция встречается примерно в 70% случаев и обычно не выявляется при обычном генетическом анализе, таком как амниоцентез.
- СПВ по однородительской дисомии: еще около 30% случаев происходят, когда человек наследует две хромосомы 15 от своей матери и ни один от своего отца. Это тип наследование называется однородительской дисомией (UPD).
- СПВ по импрессии мутации: наконец, в очень небольшом проценте случаев (1-3%) небольшая генетическая мутация при синдроме Прадера — Вилли приводит к тому, что генетический материал отцовской хромосомы 15 хоть и присутствует, но неактивен.
Как эти генетические дефекты вызывают симптомы, наблюдаемые при синдроме Прадера — Вилли?
Хромосомы 15 одни из самых сложных областей генома человека. Хотя были достигнуты значительные успехи в понимании и характеристике генетических изменений, связанных с синдромом Прадера — Вилли, точный механизм, с помощью которого отсутствие функционального генетического материала приводит к симптомам, связанным с СПВ, не понятны.
Ученые активно изучают нормальную роль генетических последовательностей в области СПВ и то, как их потеря влияет на гипоталамус и другие системы организма.
Существуют ли различия в степени тяжести СПВ в зависимости от генетического подтипа?
Могут быть некоторые тонкие различия в признаках СПВ на основе генетического подтипа: например, лица с делециями могут быть светлокожими со светлыми волосами по сравнению с другими членами семьи и быть более восприимчивыми к припадкам и судорогам; а те, у кого есть проблемы с СПВ по однородительской дисомии, могут быть подвержены более высокому риску психических заболеваний в молодом подростковом возрасте.
В целом, однако, существует значительное совпадение между различными генетическими подтипами. Вероятно, что тысячи генов за пределами области СПВ, которые демонстрируют нормальные различия между индивидуумами, также вносят значительный вклад в вариабельность симптомов синдрома Прадера — Вилли между теми, у кого есть расстройство.
Как диагностируется СПВ?
Данный синдром диагностируется с помощью анализа крови, который выявляет генетические аномалии, специфичные для синдрома Прадера — Вилли — так называемый «анализ метилирования ДНК». Метод FISH (флуоресцентная гибридизация in-situ) идентифицирует синдром путем делеции, но не диагностирует другие формы СПВ.
Анализ на метилирование ДНК идентифицирует все типы этого синдрома и является предпочтительным методом исследования для диагностики. Если анализ на метилирование проводится первым, может потребоваться дополнительное исследование, чтобы определить, вызван ли СПВ отцовской делецией, UPD или импринтинговой мутацией.
В случаях, когда существует подозрение на наличие импринтинговой мутации, кровь также может быть взята у родителей.
Каковы риски синдрома Прадера — Вилли в будущем?
СПВ по исключению и UPD являются случайными явлениями и, как правило, не связаны с повышенным риском рецидива в будущих беременностях. В случае импринтинг-мутации синдром Прадера — Вилли может в семье повториться. Семьи с опасениями по поводу их риска СПВ должны поговорить с генетическим консультантом.
Есть ли лекарство от синдрома Прадера — Вилли?
В настоящее время нет никакого лечения от синдрома Прадера — Вилли, и большинство исследований на сегодняшний день было направлено на лечение определенных симптомов данного синдрома.
Для многих людей, затронутых этим расстройством, устранение некоторых из самых сложных аспектов синдрома, таких как ненасытный аппетит и ожирение, будет представлять собой значительное улучшение качества жизни и способности жить независимо.
Источник
Синдром Прадера-Вилли (СПВ) – этиология, клиника, диагностикаСиндром Прадера-Вилли (СПВ) характеризуется перееданием, избыточным весом и рядом других соматических и поведенческих проявлений. Заболевание встречается с частотой около 1 на 7000 детей, доживших до одного года (Akefeldt et al., 1991). Распространенность синдрома среди мальчиков и девочек примерно одинакова. а) Патогенез. Синдром Прадера-Вилли (СПВ) вызван отсутствием на отцовской ДНК хромосомного локуса 15q11. Возможна делеция отцовского гена или дисомия по материнской линии. В ходе одного из исследований было выявлено, что отцы предварительно контактировали с бензолом (Akefeldt et al., 1995). Примерно в половине случаев синдром связан с интерстициальной хромосомной делецией 15q11—13 (Ledbetter et al., 1982; Mattei et al., 1983; Caldwell и Taylor, 1988). Практически во всех случаях делеция имеет отцовское происхождение (Butler et al., 1986; Knoll et al., 1989). Приблизительно в 40% случаев, без цитогенетических делеций, делеции выявляются при исследовании ДНК, а в 60% случаев отмечается дисомия всей или части 15-й хромосомы матери (Mascari et al., 1992). СПВ и синдром Ангельмана являются классическими образцами импринтинга, то есть различного экспрессирования гена в зависимости от пола родителя, передавшего ген. Ген синдрома Прадера-Вилли экспрессируется только при наследовании от отца (Wevrick et al., 1994). В случаях, когда не выявлено аномалий 15-й хромосомы, необходимо повторное клиническое обследование для выявления другого заболевания, такого как синдром Смита-Магениса (Greenberg et al., 1991). Пациенты с делецией имеют более типичные проявления, чем при ее отсутствии (Mattei et al., 1983), хотя атипичные формы заболевания отмечались и в случаях подтвержденной делеции 15q (Pauli et al., 1983; Schwartz et al., 1985; Miike et al., 1988). В рамках исследования было проведено сравнение 21 ребенка с делецией и 18 детей с нормальным хромосомным набором с высокой разрешающей способностью (Butler et al., 1986). Различия между двумя группами были выражены минимально. Чуть больше половины пациентов с делецией (11 из 21) ходили к 24 месяцам (по сравнению с двумя третями пациентов без делеции). У детей с делецией отмечались более светлые волосы, чем у детей без нее, и у всех были голубые глаза, в то время как только 13 из 18 детей без делеции были голубоглазыми (Lee et al., 1994).
б) Диагностика. Клинический диагноз синдрома Прадера-Вилли (СПВ) подтверждается типичными изменениями ДНК, выявляемыми при анализе на ген синдрома Прадера-Вилли на 15-й хромосоме FISH-методом. В период новорожденности данное заболевание следует заподозрить в случае низкой массы тела при рождении, заметной гипотонии, нарушения глотания, требующего кормления через зонд, и дисморфизма. В более старшем возрасте признаками заболевания могут быть ожирение и аномалии лица. Следует проводить дифференциальную диагностику СПВ и нейромышечных заболеваний и гипотонии, связанных с заболеваниями мозга. Необходимо рассматривать врожденную мышечную дистрофию, миотоническую дистрофию новорожденных и врожденные миопатии. Тем не менее, при СПВ на первый план выступает дисморфизм, а также нарушения сосания и глотания без значимых нарушений дыхания. В более старшем возрасте может вставать вопрос о других причинах гипотонии мозгового происхождения. Результаты биопсии мышц (которой следует избегать), уровень креатинкиназы крови и показатели электромиограммы (ЭМГ) не выходят за пределы нормы. Синдром Коэна (Norio et al., 1984) может имитировать синдром Прадера-Вилли и передается аутосомно-рецессивным путем. Особую диагностическую значимость имеют аномалии глаз (хориоретинальная дистрофия, атрофия зрительного нерва). Признаком данного заболевания являются аномалии зубов. Возможен широкий разброс клинических проявлений отдельных случаев, включая слабовыраженные формы. У многих детей могут отсутствовать выраженные трудности в обучении. Таким образом, диагноз синдрома Прадера-Вилли стоит рассматривать во всех случаях «чрезвычайно толстых и голодных» детей с проявлениями поведенческого фенотипа. в) Клинические проявления. Клинические проявления синдрома очень характерны, особенно в период новорожденности; при осмотре в более позднем возрасте большую роль при постановке диагноза играет типичный неонатальный анамнез. Новорожденные с синдромом Прадера-Вилли обычно рождаются в срок, но масса тела не соответствует гестационному возрасту. 30% детей рождаются в ягодичном предлежании. С рождения у пациентов отмечается тяжелая мышечная гипотония, поэтому часто предполагается наличие мышечного заболевания. Нарушения глотания требуют питания через зонд в течение 2-5 недель. Дисморфические проявления соответствуют кариотипу, определение которого требуется практически во всех случаях (Stephenson, 1980). В дальнейшем клиническая картина меняется. Наиболее характерными признаками являются гиперфагия и ожирение, развивающиеся у большинства детей к концу второго года жизни. Для поздней стадии заболевания характерно выраженное ожирение, стойкая гипотония и часто слабость проксимальных отделов и относительно слабо выраженная задержка умственного развития со средним коэффициентом IQ 65 (Butler et al., 1986), хотя до 10% взрослых пациентов могут иметь нормальный коэффициент IQ (Clarke et al, 1989; Greenswag, 1987). К частым проявлениям относятся также гипогонадизм, крипторхизм, низкорослость и маленький размер лица, носа, ушей, кистей и стоп (Holm et al., 1993). Часто встречается характерный внешний вид с маленькой «круглой» головой и миндалевидными глазами. Взрослые с синдромом Прадера-Вилли характеризуются низким ростом, избыточной массой тела, когнитивными нарушениями и эмоциональной лабильностью. У многих пациентов отмечается кифоз, сколиоз и остеопороз. Слабо развиты двигательные навыки, повышен аппетит (Greenswag, 1987; Lee, 2002) и имеется характерный поведенческий фенотип, включающий дерматилломанию и другие варианты поведения со склонностью к повторяющимся действиям (Akefeldt et al., 1991). В атипичных случаях возможна задержка умственного развития тяжелой степени (3%) или нормальные когнитивные способности. Иногда у пациентов может не быть ожирения, а в редких случаях отмечалась кахексия (Miike et al, 1988).
Результаты недавних исследований свидетельствуют о том, что задержка умственного развития не является обязательным проявлением (Clarke et al., 1989), и зарегистрированы случаи микроделеции 15ql 1, сопровождающиеся всеми проявлениями синдрома за исключением задержки умственного развития, низкого роста и маленьких стоп и кистей (Akefeldt et al., 1991). В настоящее время считается, что СГ1В с классически описанными симптомами может являться частью фенотипа, включающего определенные поведенческие проявления с дебютом на первом году жизни. Если ребенок начинает толстеть на втором или третьем году жизни, родители часто обращаются за помощью. В это время ребенок может быть необычно послушным или спокойным, но быть подверженным эпизодам раздражительности и чрезвычайно тяжелым и деструктивным приступам гнева. Гнев обычно немедленно сменяется «исходной» пониженной активностью и угрызениями совести. Результаты недавних исследований позволяют предположить, что обсессивно-компульсивное поведение иногда чрезвычайно напоминает расстройства аутистического спектра, но в ходе контрольных исследований не удалось выявить превышающую ожидаемую частоту расстройств аутистического спектра, принимая во внимание степень общей неспособности к обучению (Descheemaeker et al., 2006). Большинство детей с синдромом Прадера-Вилли обладает странной привычкой ковырять кожу и наносить себе раны, синяки и царапины. Большая часть детей с синдромом Прадера-Вилли страдает булимией во время обострения. Они готовы на что угодно для получения доступа к еде в холодильнике, морозилке или кладовке. Большинство родителей вынуждено применять замки для ограничения доступа детей к еде. Даже в ходе научных исследований была наглядно продемонстрирована неспособность данных пациентов прекратить есть (в экспериментальных условиях). г) Лечение синдрома Прадера-Вилли. Следует информировать родителей о природе состояния их детей. Во всех случаях необходимы беседы и печатные пособия. Врачи, диетологи, психологи (и сами родители) часто винят родителей в выраженном ожирении детей. Сопутствующие нарушения поведения часто приводят к направлению пациентов в детскую психиатрическую службу. Без подтвержденного диагноза существует большой риск, что консультирование будет сконцентрировано на воспитании как основе имеющихся отклонений. Положительное психологическое влияние на родителей оказывает информация о практически идентичных поведенческих проблемах у других детей с синдромом Прадера-Вилли. Важным моментом является обращение родителей в национальную или региональную группу поддержки больных с синдромом Прадера-Вилли. Следует посоветовать родителям (хотя большинство из них начинают делать это самостоятельно) запирать холодильники, буфеты и прочее. При очень строгом контроле со стороны родителей и сотрудников школы иногда удается заставить ребенка худеть и даже достигнуть нормального веса. Тем не менее, соблюдение данного строгого режима, безусловно, очень сложно и не следует винить семью, которая не сможет справиться со всеми ограничениями. Для снижения веса (и необычайно высокого уровня серотонина в спинномозговой жидкости) при синдроме Прадера-Вилли применялись различные диеты и препараты, включая фенфлурамин, но даже в случае временного успеха долгосрочный эффект не был достигнут. Последние 10 лет в качестве компонента полной программы лечения детей и взрослых с синдромом Прадера-Вилли начато применение гормона роста. Применение данного препарата приводит к достижению более высокого роста и некоторому улучшению умственных способностей. Тем не менее, присутствует предположение о возможности развития значимых (и иногда опасных) побочных эффектов у некоторых пациентов, что означает необходимость назначения терапии гормоном роста только врачами-специалистами, работающими в данной области. – Также рекомендуем “Синдром Ретта – этиология, клиника, диагностика” Редактор: Искандер Милевски. Дата публикации: 5.12.2018 Оглавление темы “Наследственные синдромы в неврологии.”:
|

Источник