
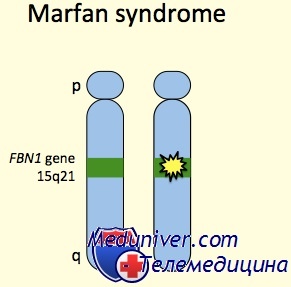
У человека синдром марфана наследуется как доминантный

Синдром Марфана: причины, диагностика, лечениеЭтиология и встречаемость синдрома Марфана. Синдром Марфана (MIM №154700) — панэтническое аутосомно-доминантное заболевание соединительной ткани, вызванное мутациями в гене фибриллина 1 (FBN1, MIM № 134797). Синдром Марфана имеет встречаемость около 1 на 10 000. Приблизительно 25-35% пациентов имеют новую мутацию. Мутации, вызывающие синдром Марфана, разбросаны по гену, и каждая мутация обычно уникальна в семье. Патогенез синдрома МарфанаГен FBN1 кодирует фибриллин 1, внеклеточный матричный гликопротеид с широким распределением. Фибриллин 1 полимеризуется, формируя микрофибриллы как в эластичных, так и в неэластичных тканях, например стенке аорты, цилиарных поясках и коже. Мутации влияют на синтез, процессинг, секрецию, полимеризацию или устойчивость фибриллина 1. Исследования накопления и экспрессии фибриллина 1 в культуре клеток предположили доминантный отрицательный патогенез, т.е. синтез мутантного фибриллина 1 тормозит образование нормальных микрофибрилл нормальным фибриллином 1 или стимулирует протеолиз несоответствующих внеклеточных микрофибрилл. Последние исследования на мышиных моделях синдрома Марфана указывают, что половинного количества нормального фибриллина 1 недостаточно, чтобы проводить эффективную сборку микрофибрилл. Таким образом, патогенезу болезни также может содействовать гаплонедостаточность. Кроме синдрома Марфана, мутации в гене FBN1 могут вызывать другие синдромы, включая неонатальный синдром Марфана, изолированные скелетные симптомы, аутосомно-доминантную эктопию хрусталиков и фенотип MASS (марфаноидные симптомы, включая пролапс митрального клапана или миопию, пограничное и непрогресирующее расширение аорты, и неспецифические изменения скелета и кожи). В общем, фенотипы довольно схожи в пределах семьи, хотя тяжесть фенотипических проявлений может значительно изменяться. До настоящего времени точное соотношение между генотипом и фенотипом не определено. Внутрисемейная и межсемейная изменчивость позволяет предполагать, что в определении фенотипа важную роль играют окружающая среда и эпигенетические факторы. Последние исследования на мышиных моделях показывают, что фибриллин 1 не просто структурный белок, и что синдром Марфана не просто результат структурной слабости тканей. Более того, микрофибриллы фибриллина 1 в норме связывают и уменьшают концентрацию и активность факторов роста суперсемейства TGFb. Потеря фибриллина 1 увеличивает сигналы свободного TGFb значительно содействующие заболеванию, так как антагонисты TGFb устраняют легочные и клапанные изменения, наблюдаемые у мышей с недостаточностью фибриллина 1.
Фенотип и развитие синдрома МарфанаСиндром Марфана — мультисистемное заболевание со скелетными, глазными, сердечно-сосудистыми, легочными, кожными и другими аномалиями. Скелетные аномалии включают очень высокий рост (отношение размаха рук к росту >1,05; соотношение верхнего и нижнего сегментов <0,85 у взрослых), арахнодактилию, аномалии грудины, сколиоз, разболтанность суставов, готическое нёбо. Аномалии глаз включают подвывих хрусталиков, уплощение роговицы, удлинение глазного яблока и гипоплазию радужки. Сердечнососудистые аномалии включают пролапс митрального клапана, аортальную регургитацию и расширение и расслаивающую аневризму восходящей аорты. Легочные аномалии включают спонтанный пневмоторакс и расширение концевых пузырьков. Аномалии кожи включают атрофические бороздки и рецидивирующие грыжи. Аномалии твердой мозговой оболочки включают выбухание оболочки в крестцово-поясничном отделе. Большинство признаков синдрома Марфана появляются с возрастом. Скелетные аномалии типа аномалии грудины и сколиоза ухудшаются с ростом костей. Подвывих хрусталика часто присутствует уже в раннем детстве, но может развиваться и в юности. С повышенной частотой при синдроме Марфана встречаются отслойка сетчатки, глаукома и катаракты. Сердечно-сосудистые осложнения обнаруживаются в любом возрасте и развиваются в течение всей жизни. Основные причины преждевременной смерти пациентов с синдромом Марфана — сердечная недостаточность вследствие регургитации клапанов и аневризмы и разрыва аорты. Тем не менее в связи с улучшением хирургической и терапевтической помощи при аневризме аорты выживание улучшилось. С 1972 по 1993 г. ожидаемый возраст выживания для 50% пациентов поднялся с 49 до 74 лет для женщин и с 41 до 70 лет для мужчин. Особенности фенотипических проявлений синдрома Марфана:
Лечение синдрома МарфанаСиндром Марфана — клинический диагноз, определяемый по наличию конкретных симптомов. Подтверждение синдрома Марфана идентификацией мутаций в гене FBN1 в настоящее время практически нецелесообразно, поскольку крайняя аллельная гетерогенность делает идентификацию причинно-обусловленной мутации в каждой семье крайне трудозатратной, а также из-за недостаточно надежной корреляции между генотипом и фенотипом. Анализ мутаций имеет недостаточную чувствительность или специфичность для синдрома Марфана, что ограничивает его клиническую пользу. Для синдрома Марфана недоступно эффективное лечение; следовательно, помощь сфокусирована на профилактике осложнений и симптоматическом лечении. Оказание офтальмологической помощи включает регулярные осмотры, коррекцию миопии и, часто, замену хрусталика. Ортопедическая помощь заключается в укрепляющем лечении или хирургической коррекции сколиоза. Помощь при аномалии грудины в основном косметическая. Физиотерапия может компенсировать нестабильность суставов. Сердечно-сосудистые проблемы решаются комбинацией терапевтических и хирургических мероприятий. Терапевтические усилия направлены на предохранение или замедление развития расширения корня аорты за счет уменьшения кардиологических показателей, снижения артериального давления и усилия выброса желудочков с помощью бета-адреноблокаторов, ограничение участия в контактных видах спорта, соревновательных видах спорта и в изометрических упражнениях. Профилактическая замена корня аорты показана, когда расширение аорты или аортальная регургитация становится достаточно тяжелой. Большинству пациентов в настоящее время проводят надклапанную замену корня аорты, не требующую постоянного приема противосвертывающих препаратов. Гемодинамические изменения, связанные с беременностью, могут приводить к прогрессирующему расширению или расслоению аорты. Полагают, что расслоение аорты вторично к гормональным изменениям, увеличению объема крови и сердечного выброса, связанных с беременностью и родами. Современные исследования считают, что риск беременности слишком велик, если ширина корня аорты превышает 4 см. Женщины могут выбрать проведение надклапанной замены аорты перед беременностью. Риски наследования синдрома МарфанаПациенты с синдромом Марфана имеют 50% риск иметь ребенка, больного синдромом Марфана. В семьях, передающих синдром Марфана, членов семьи, находящихся в группе риска, можно выявлять, либо обнаруживая мутацию (в тех редких случаях, когда она известна), либо анализом сцепления, если маркеры, тесно сцепленные с локусом FBN1, имеют очевидную связь с болезнью в семье пробанда. Пренатальная диагностика доступна только для семей, в которых возможны исследования сцепления или известна мутация в гене FBN1. Пример синдрома Марфана. Д.Л., здоровый 16-летний ученик средней школы, звезда баскетбола, направлен в генетическую клинику для обследования по поводу синдрома Марфана. Телосложение Д.Л. похоже на телосложение его отца. Отец Д.Л., высокий субтильный человек, умер во время утренней пробежки; у других членов семьи случаев скелетных аномалий, внезапной смерти, снижения зрения или врожденных аномалий не было. При медицинском осмотре выявлены астеническое телосложение, высокое дугообразное нёбо, небольшая деформация грудины по типу «куриной» груди, арахнодактилия, соотношение размах рук/рост 1,1, диастолический шум и стрии на плечах и бедрах. Эхокардиография выявила расширение корня аорты с аортальной регургитацией. Офтальмологическое обследование показало двусторонний иридодонез и легкое смещение хрусталиков кверху. На основе медицинского осмотра и результатов обследования генетик объяснил пациенту и его матери, что у него синдром Марфана. – Также рекомендуем “Синдром Миллера-Дикера: причины, диагностика, лечение” Оглавление темы “Наследственные болезни”:
|
Источник

Синдром Марфана – заболевание соединительных тканей, при котором нарушается синтез белка фибриллина, важной составляющей многих структур организма. Встречается с частотой 1 случай на 10-20 тысяч человек, при этом раса и пол не имеют значения. В 75% случаях синдром Марфана наследуется от родителей, а в остальных – является результатом мутаций.
Продолжительность жизни
В отсутствии лечения больные с синдромом Марфана редко проживают больше 30-40 лет, однако при своевременном обнаружении заболевания и выполнении рекомендаций врача срок жизни можно продлить. На сегодняшний день люди с синдромом Марфана могут доживать до преклонного возраста, ведут умеренно активный образ жизни. Распространенная причина смертности пациентов – разрыв аорты, который происходит при интенсивных физических нагрузках.
В этом заключается наибольшая опасность заболевания – если его не диагностировали вовремя, человек считает себя абсолютно здоровым и может подвергать себя перенапряжению во время бега на спринтерские дистанции, плавания, езды на велосипеде либо заниматься тяжелым физическим трудом. Ввиду того, что люди с синдромом Марфана зачастую высокие с длинными конечностями, имеют большой размах рук, им часто рекомендуют попробовать себя в спорте, что повышает риск смертности.
Содержание:
- Симптомы синдрома Марфана
- Причины синдрома Марфана
- Диагностика синдрома Марфана
- Лечение синдрома Марфана
Симптомы синдрома Марфана
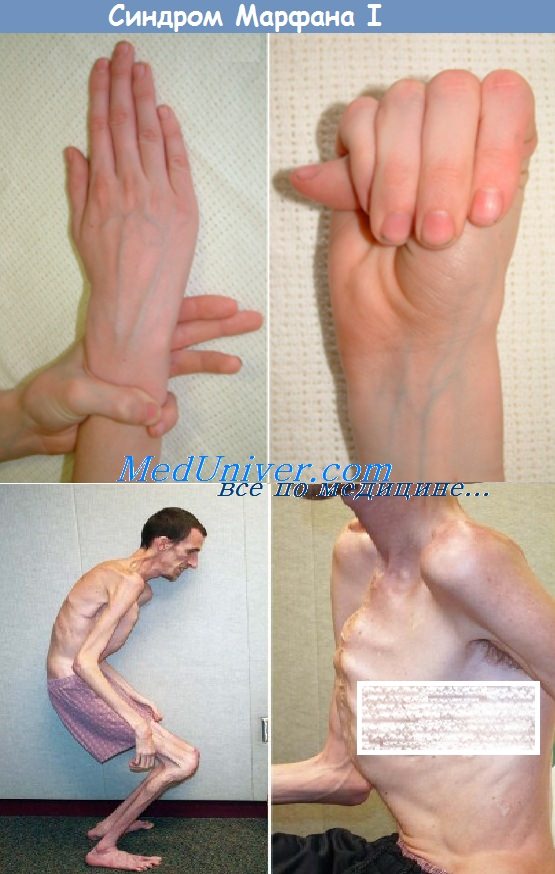
Пациенты с синдромом Марфана имеют специфическую внешность, что упрощает диагностику заболевания. Обычно они имеют высокий рост, удлиненные конечности, при этом нижняя часть туловища длиннее верхней, а размах рук больше, чем рост. Астеничное телосложение сочетается с недостаточной массой тела, которая часто меньше 50 кг. Удлиненные пальцы рук (арахнодактилия или «паучьи пальцы») позволяют диагностировать заболевания по симптому запястья и симптому большого пальца (когда палец выглядывает на целую фалангу из руки, даже если зажать его в кулак).
Людей с синдромом Марфана отличают характерные черты лица, что придает ему общее для всех пациентов данной группы «птичье» выражение. Глаза глубоко посажены, череп удлиненный, зубы скученны из-за малых размеров челюсти, готическое «небо». Нос кажется крупным, уши довольно большие и низко расположенные. У одного человека редко можно встретить все эти проявления, чаще всего среди внешних признаков отмечают только высокий рост и удлиненные пальцы.
Из-за слабости соединительных тканей у пациентов с синдромом Марфана могут наблюдаться стрии (растяжки на коже), которые не связаны с резкими скачками в весе или гормональными расстройствами. Эти симптомы проявляются обычно в области нижней трети спины, поясничного отдела и в зоне плеч.
Другой часто встречающийся симптом – грыжи, которые возникают самопроизвольно либо после операции, развиваясь из-за слабости соединительных тканей. Даже после хирургического лечения грыжа у больного с синдромом Марфана может появиться вновь.
Распространенный симптом со стороны органов зрения – подвывих хрусталика, который развивается из-за патологий соединительнотканных связок, а также миопия. У пациентов может наблюдаться отслоение сетчатки, вторичная глаукома, катаракта и другие глазные заболевания. Потеря зрения наряду с сердечнососудистыми патологиями является главной опасностью синдрома Марфана.
Со стороны опорно-двигательного аппарата часто наблюдаются искривления позвоночника по типу лордоза, кифоза и сколиоза, вдавливание и деформация грудной клетки, гиперподвижность суставов, гипотония мышц и плоскостопие.
Со стороны дыхательной системы часто появляются патологии, связанные с искривлением грудной клетки, в результате которого она сдавливает легкие. Повышен риск спонтанного пневмоторакса, что следует учитывать при занятиях спортом и физической активностью.
Со стороны сердечнососудистой системы наблюдают расслоение стенок аорты, патологии сердечных клапанов.
Что касается интеллектуального развития пациентов с синдромом Марфана, то оно не опускается ниже границ нормы, а в некоторых случаях может значительно превышать ее. Так, при среднем коэффициенте интеллекта в 85 единиц у больных с синдромом МарфанаIQ может быть больше 115, что является очень высоким показателем и встречается у одаренных людей. Считается, что это связано с высоким уровнем адреналина у лиц с данным заболеванием, из-за чего они амбициозны, стремятся быть лучше других по многим показателям и быстрее усваивают информацию.
Психические процессы при синдроме Марфана протекают своеобразно – дети с данным заболеванием могут быть гиперактивными, нервными и раздражительными, обладают завышенной самооценкой, болезненно реагируют на малейшие раздражители, часто плачут. Однако все эти проявления не указывают однозначно на наличие заболевания, а могут быть связаны с особенностями темперамента и протекания психических процессов.
Симптомы, связанные с нарушениями зрения проявляются довольно рано – подвывих хрусталика обнаруживают даже у годовалых детей или диагностируют в 6-7 лет перед поступлением в школу. Другие патологии зрения – глаукома, отслоение сетчатки и катаракта – появляются у пациентов в возрасте от 15 до 40 лет.
Синдром Марфана проявляется чувством сильной усталости и истощения после физических нагрузок, боль в мышцах, слабость и гипотония – все это свидетельствует о нарушении функции митохондрий, которые обеспечивают энергетические процессы в клетках.
Причины синдрома Марфана

Синдром Марфана – генетическое заболевание с наследуемостью аутосомно-доминантного типа, причиной его развития являются мутации гена FBN1. Этот ген отвечает за выработку фибриллина-1 – важного структурного белка, входящего в состав связок и эластичных сосудов. При нарушениях в строении этого белка соединительнотканные структуры становятся более растяжимыми, при этом теряют устойчивость к деформациям. Наиболее опасные последствия таких изменений проявляются со стороны органов зрения и сердечнососудистой системы. Фибриллин-1 необходим для нормального функционирования цинновой связки, прикрепляющей хрусталик глаза к цилиарному телу. Поэтому при нарушении его синтеза, что происходит у больных с синдромом Марфана, цинновая связка ослабляется, что на первых этапах проявляется близорукостью у пациента, подвывихом хрусталика, но в дальнейшем может привести к вторичной глаукоме, частичной или полной потере зрения.
Другие функции фибриллина в организме – формирование внеклеточного матрикса, что обеспечивает целостность соединительных тканей и нормальное функционирование факторов роста клеток.
Аорта относится к сосудам эластического типа и содержит множество эластических связок, от которых зависит ее устойчивость к нагрузкам. При нарушении их функции возникает расширение аорты, расслоение ее стенок. Такая ситуация ставит под угрозу жизнь пациента, может спровоцировать внезапную смертность из-за разрыва аорты во время интенсивных физических нагрузок. Расширение аорты опасно для женщин в период беременности, особенно это касается третьего триместра, а также во время родов и несколько месяцев после них.
Среди других распространенных сердечных патологий, связанных с синдромом Марфана – поражения митрального клапана, из-за чего возникает необходимость хирургического лечения.
Синдром Марфана наследуется?
Синдром Марфана – генетическое заболевание, которое в 75% случаях наследуется по семейному типу, а оставшиеся 25% встречаемости являются результатом первичной мутации гена. В семьях, где отец старше 35 лет риск рождения ребенка с синдромом Марфана повышается.
Развитие симптомов начинается уже в период перинатального развития – соединительнотканные волокна в процессе формирования теряют прочность, с чего начинаются изменения в эластических сосудах, опорно-двигательном аппарате, глазных связках, твердом небе, сердечных клапанах. Прогрессирование заболевания продолжается в постнатальный период, и длится всю жизнь пациента.
Диагностика синдрома Марфана
Синдром Марфана диагностируется на основании данных физикального осмотра, семейного анамнеза, обследования у кардиолога и офтальмолога, рентгенологических исследований органов грудной клетки.
Немаловажную роль при диагностике играют такие критерии:
Наличие дефектов грудной клетки, которые требуют хирургического вмешательства;
Наличие патологических изменений тканей и органов зрительной, сердечнососудистой системы, характерных для синдрома Марфана;
Эктопия хрусталика;
Верхняя часть туловища значительно короче нижней, их соотношение больше 0,86;
Размах рук больше, чем рост ( их соотношение больше 1,05);
При синдроме Марфана в несколько раз возрастает выведение глюкозаминов через почки, что также используется при диагностике.
ЭКГ и эхокардиография позволяют обнаружить нарушения сердечной деятельности. Для обнаружения патологий со стороны дыхательной системы проводят обследование внешнего дыхания. У пациентов с синдромом Марфана часто наблюдается вздутие легочных тканей, гиперкапния, неравномерное распределение воздуха по легким и разнообразные нарушения механики дыхания.
Исследования ДНК (прямое автоматическое секвентирование) позволяют определить наличие мутаций в гене FBN1, идентифицировать их.
Лечение синдрома Марфана
Лечение синдрома Марфана имеет симптоматический характер и направлено на уменьшение негативных проявлений заболевания со стороны сердечнососудистой, зрительной и других систем.
С целью профилактики осложнений больным рекомендуется избегать видов спорта, связанных с резкими бросками и рывками, как баскетбол, бега на спринтерские дистанции, контактных видов спорта, во время которых возможны телесные повреждения. Тем не менее, не следует отказываться от спорта вовсе, в группу риска попадают такие занятия, которые вызывают резкое изменение сердечного ритма, частоты сердечных сокращений. Если контролировать свой пульс во время занятий спортом и не выходить за рамки безопасных показателей, то такая активность не противопоказана.
С раннего возраста рекомендуется принимать лекарственные препараты, снижающие нагрузку на аорту, как бета-адреноблокаторы. В составе комплексного лечения детям с синдромом Марфана прописывают витаминные комплексы, антиоксиданты и энерготропные средства курсами продолжительностью в месяц по несколько раз в году.
К ним относятся:
Токоферол, аскорбиновая кислота, витамины группы B;
Рибоксин в таблетках;
КоэнзимQ10;
Элькар;
Лимонтар курсом в десять дней из расчета 5 мг на 1 кг веса;
Димефосфон.
Из ноотропных препаратов назначают пирацетам по 400 мг дважды в сутки курсом в два месяца, повторяют 3-4 раза в год.
Хирургическое вмешательство при кардиальных симптомах заболевания требуется в случае, если корень аорты у пациента достигает 5 см и больше.
В течение всей жизни больной с синдромом Марфана должен постоянно проходить обследования у специалистов различного профиля – офтальмологов, кардиологов, ортопедов и генетиков, так как заболевание проявляет себя по-разному в каждом отдельном случае, провоцируя нарушения функции различных органов и систем.
Кроме того, необходимо санировать очаги инфекции в ротовой полости с применением антибиотиков, так как у больных с синдромом Марфана ослабленный иммунитет. Антибиотикотерапию в целях профилактики используют даже при незначительных хирургических вмешательствах, чтобы снизить вероятность развития инфекционного эндокардита.
Хирургической коррекции подлежат не только патологии сердца, расслоение стенок аорты и поражения клапанов, но и заболевания внутренних органов и скелета, возникающие в ходе прогрессирования синдрома Марфана.
Бета-адреноблокаторы применяются не только в целях снижения нагрузки на сердечнососудистую систему, но и эффективно убирают негативные проявления артериальной гипертензии и аритмии, характерные для данного заболевания.
Автор статьи: Мочалов Павел Александрович | д. м. н. терапевт
Образование:
Московский медицинский институт им. И. М. Сеченова, специальность – “Лечебное дело” в 1991 году, в 1993 году “Профессиональные болезни”, в 1996 году “Терапия”.
Наши авторы
Источник